Keywords
Donor-derived cell-free DNA, liver transplantation, immunosuppression, rejection, liver biopsy
This article is included in the Genomics and Genetics gateway.
Donor-derived cell-free DNA, liver transplantation, immunosuppression, rejection, liver biopsy
Despite advancements in immunosuppressive therapies, acute cellular rejection still occurs in 10-40% of liver transplant patients. Although liver biopsy is the gold standard for diagnosing rejection, it is invasive, costly, associated with complications, causes discomfort, and has a long turnaround time. Immediately following transplantation, biopsies may be challenging due to low platelet counts, coagulopathy, and the presence of ascites.1 Moreover, conventional liver function tests and biomarkers such as bilirubin and liver enzymes are nonspecific for rejection, as they can be influenced by various factors such as sepsis and biliovascular complications, highlighting an urgent need for noninvasive, reliable tools to monitor graft rejection and tailor immunosuppressive therapy.2
Donor-derived cell-free DNA (dd-cfDNA) has emerged as a promising noninvasive biomarker for monitoring liver transplant rejection. These small fragments of DNA, released from necrotic and apoptotic donor cells, increase in the bloodstream after transplantation and stabilize within a week. During rejection episodes, dd-cfDNA levels can rise to fivefold, allowing for early detection approximately 1 to 2 weeks before clinical symptoms or biochemical abnormalities manifest.3 With a short half-life of 1.5 hours, dd-cfDNA provides real-time insights into graft health and requires only a blood sample, with a turnaround time of one day.4,5 Serial monitoring of dd-cfDNA can assess treatment efficacy, as persistent elevation indicates ongoing graft injury. Elevated dd-cfDNA on day 7 post-transplant is particularly useful for predicting rejection risk and differentiating rejection from other confounding conditions, thereby improving post-transplant prognostication and management.6
Previous research on donor-derived cell-free DNA (dd-cfDNA) in liver transplantation reveals promise and limitations. Kanamori et al.'s study across eight transplant centers in Japan (involving 19 patients) found that dd-cfDNA levels correlated with rejection activity but had limited efficacy in detecting sub-clinical rejection.7 Schütz et al.'s multicenter study in Germany (with 88 patients), demonstrated that dd-cfDNA had higher sensitivity and specificity for detecting graft injury compared to liver function tests (LFTs); however, it lacked time-matched samples of LFT and dd-cf DNA levels, and protocolized patient care.8 Baumann et al.'s larger cohort study (with 108 patients) at Hannover Medical School showed increased dd-cfDNA levels with graft rejection, but its accuracy in predicting sub-clinical damage was still limited, and no correlation with donor-specific antibodies was observed.9 These findings underscore the need to validate the diagnostic reliability of dd-cfDNA and its clinical utility in guiding immunosuppression modulation to prevent post-transplant rejection.
This study is particularly relevant for various reasons. Firstly, it is the first randomized controlled trial that explores the potential of dd-cfDNA levels to preemptively modulate immunosuppression levels, an area that has not been investigated in the past. This study will thus analyze whether dd-cfDNA-directed immunosuppression can prevent rejection. Secondly, there will be a comparative assessment of liver integrity and morphology at one-year post-transplant, using Fibroscan, or CT-MRI, to assess whether dd-cfDNA modulated immunosuppression may have long-term beneficial effects. Thirdly, this study will analyze the integrity ratio of short and long DNA fragments, percentage of dd-cfDNA specific to allograft, and total dd-cfDNA to differentiate from other potential causes of raised dd-cfDNA levels as well as to correlate with types of rejection. Finally, it aims to establish optimal thresholds and ideal sampling frequencies for cfDNA, which remain poorly defined in the current literature.
Harm & Benefits: There is no harm at all, as the study involves only blood samples which will anyway be performed during the routine follow-up as part of standard protocol. The benefits will include avoidance of invasive liver biopsy if the study proves the benefit of dd-cfDNA in the diagnosis of rejection. Furthermore, early detection may aid in the prevention of graft cellular damage by preemptive immunosuppression modulation.
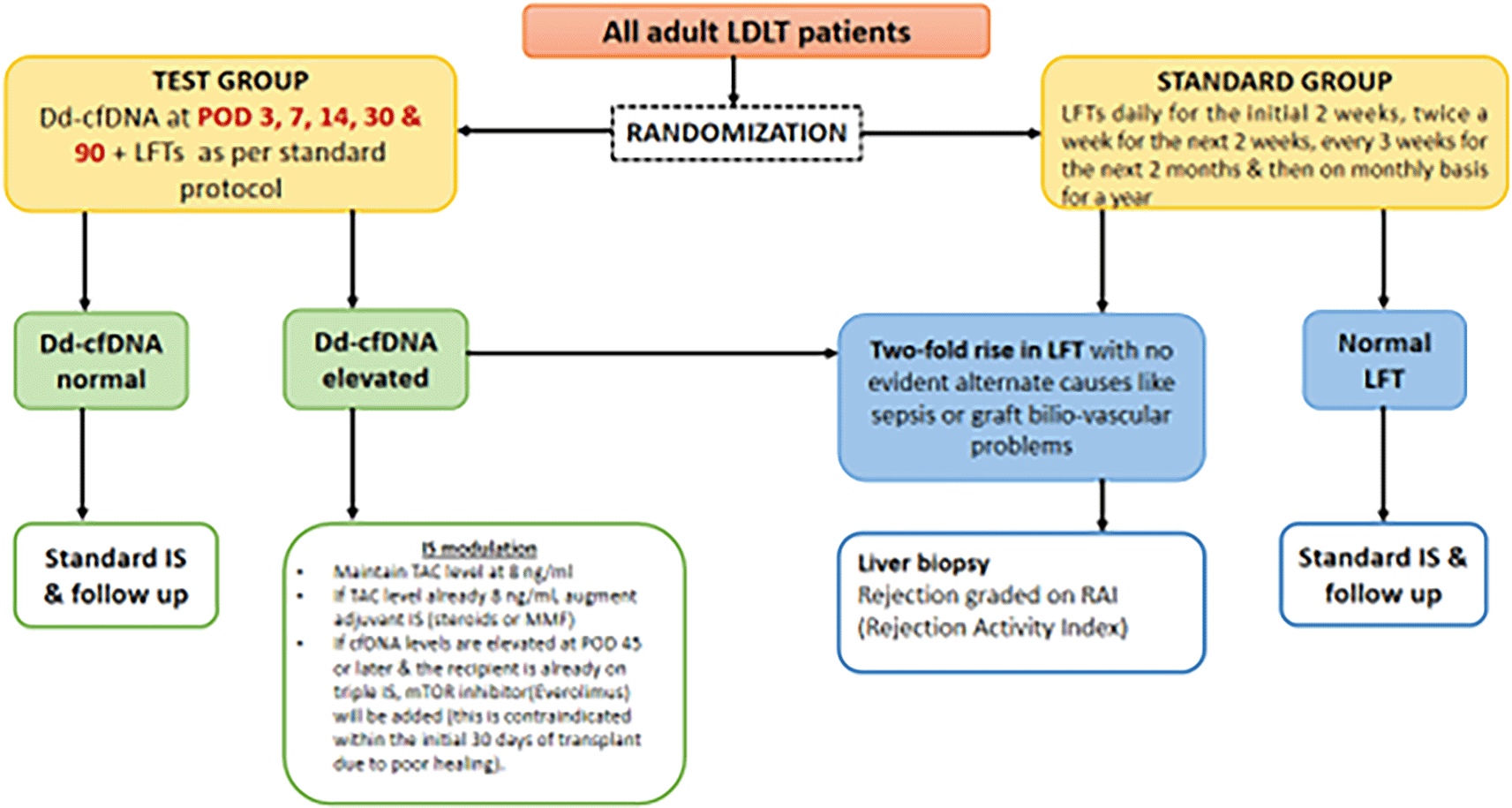
The study compares a test group and a standard group in liver transplant patients. The standard group follows protocolized care, adjusting immunosuppression based on trough levels, and clinical, and biochemical parameters. The test group undergoes additional monitoring with dd-cfDNA, allowing earlier immunosuppression adjustments. In both groups, liver biopsies for rejection diagnosis are based on LFTs. This approach in the test group may enable earlier intervention, potentially reducing rejection rates.
• Primary: To assess the efficacy of dd-cfDNA modulated immunosuppression in ameliorating acute T-cell mediated rejection following liver transplant.
• Secondary:
a) To evaluate and compare the efficacy of donor-derived cell-free DNA (dd-cfDNA) as an early biomarker for detecting allograft rejection in liver transplant recipients, in comparison to conventional liver function tests
b) To evaluate the graft morphology and stiffness by fibroscan and cross-sectional imaging at one-year after transplantation.
Trial design - Randomized controlled trial, Allocation ratio-1:1, Framework- Non-inferiority
Patient and Public Involvement (PPI) statement
Patients were not involved during the initial planning of the research questions or the methodology, but their concerns about the associated risks, discomfort, and cost of liver biopsies shaped the research focus on noninvasive alternatives. However, informed video consent was obtained from all participants before including them in the study.
• Inclusion criteria: All adult patients undergoing live donor liver transplantation
• Exclusion criteria: Multiorgan transplant patients, identical twin & donor recipients, and pediatric allograft recipients, ABO-incompatible transplantation, retransplantation
• Intervention: Use of donor-derived cell-free DNA (dd-cfDNA) to monitor liver transplant recipients for early signs of rejection and modulate immunosuppression accordingly.
• Outcome:
• Compare biopsy-proven rejection rates between the two groups (using Banff Criteria).
• Compare biochemical rejection rates, defined as a two-fold transaminase rise without alternative causes, resolving with pulsed steroids.
• Compare steroid-resistant rejection rates, confirmed by biopsy and treated with ATG (Antithymocyte Globulin)
• Assess the sensitivity and specificity of dd-cfDNA, using biopsy as the gold standard.
• Evaluate whether dd-cfDNA-modulated immunosuppression better preserves graft integrity at one year.
• Correlate antibody-mediated rejection with Granzyme-B levels.
Study setting - Conducted at Amrita Institute of Medical Sciences and Research, Kochi, Kerala, India (single-center study)
A. Interventions for each group
Post liver transplantation, both groups of patients will initially be started on immunosuppression as follows:
▪ Tacrolimus will be started at 2mg/day either in divided or single doses depending on the formulation to aim for a trough level of 8ng/ml. The dosage will be altered depending on the renal function.
▪ Methylprednisolone will be started at 250 mg IV on Day 1, 125 mg IV on Day 2, 80 mg IV on Day 3, and 40 mg IV on Day 4. Subsequently, oral prednisolone will be started at 30mg daily tapered weekly by 5mg, and stopped at 6 weeks.
▪ Mycophenolate Mofetil (MMF) will be started at 500 mg twice daily from day 1, provided the platelet count is more than 25,000/mm3. It will be stopped at 4-6 months. Due to the complexity of the interaction between immunosuppressants and other drugs, as well as hepatic and renal function, some degree of immunosuppression “tweaking” will be left to the physician's discretion.
▪ On postoperative day 3, the participants will be randomly assigned into two groups using a computerized randomization technique, with the assigned group enclosed in sealed, opaque envelopes opened only by the study coordinator.
▪ Group 1 (Test group): Blood samples (9ml) will be collected on the postoperative days 3,7,14,30 & 90 and the dd-cfDNA will be quantified. At any point during the analysis, if dd-cfDNA levels are found to be elevated, this possibly suggests an early rejection and immunosuppression will be adjusted as follows:
▪ The primary goal of immunosuppression modulation is to maintain Tacrolimus (TAC) blood levels at 8 ng/ml. If TAC levels are already at this target level, immunosuppressive therapy will be intensified by either increasing the dosage or adding alternative immunosuppressants such as steroids or Mycophenolate mofetil (MMF) depending on clinical parameters. Steroid dose will be increased by 10mg of prednisolone or Mycophenolate mofetil will be increased to 500mg thrice a day. This will be based on the level of the platelet count and total WBC count.
▪ If the dd-cfDNA levels are elevated on postoperative day 30 or 90, and the recipient is already on adequate triple immunosuppression, mTOR inhibitor (Everolimus) will be added. This is a relative contraindication within the initial 30 days of transplant due to poor wound healing.
▪ Simultaneously if LFTs show significant abnormalities, (more than a two-fold rise in transaminases, alkaline phosphatase or gamma glutamyl transferase) without evident causes (like sepsis or graft-related issues such as biliovascular problems) a liver biopsy will be performed to confirm rejection and it will be graded according to the Rejection Activity Index (RAI). Biopsy-proven rejection will be treated with pulsed doses of Methylprednisolone (10 mg/kg for 3-5 days) as per the protocol of the unit.
▪ If liver biopsy is contraindicated (due to coagulopathy, ascites, logistical issues), alternative assessments will rule out sepsis and biliovascular complications using CT/MRI and sepsis markers (WBC, CRP, procalcitonin, cultures, virological assays). Patients will typically receive pulsed MethylPrednisolone at 10 mg/kg for 3 to 5 days, and a response to this treatment will lead to a provisional diagnosis of biochemical rejection.
▪ Steroid-resistant rejection is defined as a form of acute rejection in which the liver transplant recipient does not respond adequately to three aliquots of pulsed steroids This will be confirmed by liver biopsy and treated usually by Antithymocyte globulin (ATG).
▪ dd-cfDNA analysis will involve the assessment of parameters like total cfDNA, dd-cfDNA percentage, integrity ratio, and Granzyme-B to differentiate between infection, T-cell mediated rejection, antibody-mediated rejection, and normal profile. This will help in precisely identifying the presence and type of rejection in each patient.
▪ If dd-cfDNA levels are found to be normal, the patient will continue with standard immunosuppressive therapy, with regular follow-up to ensure stable transplant function.
Group 2 (Standard group): Patients are monitored using standard liver function tests alone. A biopsy will be performed if there is a two-fold rise in liver function tests (aminotransferases, alkaline phosphatase, gamma-glutamyl transferases) without evident causes like sepsis or graft-related issues (such as biliovascular problems). Biopsy-proven rejection will be treated as elaborated in Group 1.
Criteria for discontinuation- There are no criteria for discontinuation, as the study involves only collecting blood samples for dd-cfDNA analysis along with those required for follow-up lab investigations and immunosuppression modulation will be followed according to the protocol. However, there may be a requirement to change or even cease immunosuppression in case of adverse effects which will be captured in the data sheet for analysis.
B. Strategies to improve adherence to protocol- Regular follow-ups and tests, educating participants on the protocol's importance, and using reminders (SMS, calls) for appointments and medication.
D. Relevant concomitant care and interventions prohibited/permitted during the trial
a. Efficacy of dd-cfDNA in indicating adequate immunosuppression levels.
• Specific Measurement: Occurrence of biopsy-proven rejection in both groups according to Banff criteria (It is a standardized system for grading transplant rejection based on biopsy findings, assessing inflammation, bile duct damage, and endothelial injury.)
• Analysis Metric: Percentage of dd-cfDNA at various time points and Tacrolimus levels and the modus of immunosuppression changes
• Method of Aggregation: Mean of dd-cfDNA levels, integrity ratio, total cfDNA, and percentage of graft-specific cfDNA
• Time Point: Measured at postoperative days 3, 7,14,30 and 90
• Clinical Relevance: Modulation of immunosuppression by dd-cfDNA along with the standard protocol may reduce the actual number of biopsy-proven rejections.
b. Detection of biochemical rejection.
• Specific Measurement: LFTs (AST, ALT, ALP, GGT, bilirubin), cross-sectional imaging (CT/MRI), markers of sepsis (WBC, CRP, procalcitonin, bacterial cultures, virological assays)
• Analysis Metric: Two-fold rise in liver function tests with normal cross-sectional imaging, and negative markers of sepsis
• Method of Aggregation: Median with standard deviation of LFTs
• Time Point: LFTs were measured every day for 2 weeks then alternate days for 2 weeks and then weekly for 3 months. Imaging and markers of sepsis will be performed as dictated clinically.
• Clinical Relevance: To assess the rates of biochemical rejection in 2 groups
c. Graft morphology at 1 year.
• Specific Measurement: Liver integrity is assessed via CT/MRI or Fibroscan.
• Analysis Metric: Liver stiffness measurement (LSM), Controlled Attenuation Parameter (CAP)
• Method of Aggregation: Percentage of participants with intact graft function.
• Time Point: One-year post-transplantation
• Clinical Relevance: Sub-clinical graft damage at one year can be assessed to find whether it has any correlation with the number of rejection episodes.
Harm Outcomes: None
The inclusion and exclusion criteria are outlined in Figure 1, while the detailed methodology of the study is presented in Figure 2. Additionally, the SPIRIT figure, which illustrates the timeline for eligibility screening, patient enrollment, and assessment, is provided in Table 1.

Based on the proportion of rejection in the treatment group (33%) and in the control group (13%) in liver transplant patients, was observed in a pilot study conducted with 30 samples (15 in each group) and with 80% power and 95% confidence, the sample size comes to 136 with 68 in each group.
All patients undergoing live donor liver transplantation in the Department of Gastrointestinal Surgery, Amrita Institute of Medical Sciences and Research will be evaluated.
A. Sequence of allocation: The informed consent and baseline assessments will be obtained from all included patients. On postoperative day 3, the study coordinator will assign the participants randomly to either Group 1 (Test group-dd-cfDNA and LFT monitoring) or Group 2 (Standard group-LFT monitoring only) using a computerized randomization list, in a 1:1 ratio.
B. Allocation Concealment: A biostatistician will generate a computerized randomization list to ensure that those involved in recruiting and assessing participants cannot influence the allocation process. The assigned group of each patient will be enclosed in sealed, opaque envelopes opened only on postoperative day 3.
C. Implementation: Once the dd-cfDNA is analyzed for each blood sample, the results will be communicated to the healthcare team responsible for implementing the intervention. The study will be closely monitored with regular audits to confirm that participants are receiving the appropriate monitoring based on their assigned group.
A. Who will be blinded - The laboratory scientists analyzing the samples for dd-cfDNA levels will be blinded to the LFT values and the participants' clinical conditions to minimize bias. However, the study researchers cannot be blinded, as they are responsible for adjusting immunosuppression based on the dd-cfDNA levels.
B. Circumstances under which unblinding is permissible- NA
A. Collection & Assessment of Outcomes
Blood samples will be collected on designated postoperative days and during follow-up visits, with demographic and clinical data recorded on a predefined form and in Excel. To ensure data quality, the study will implement standardized protocols, conduct audits every three months, utilize dual entry methods, maintain thorough documentation, and involve a biostatistician for data analysis.
B. Plans to promote participant retention & follow-up
To ensure participants fully understand the study's purpose, procedures, and benefits, detailed information will be provided upfront. Regular follow-up visits and check-ins, via phone or messages, will help maintain engagement and monitor participants' well-being. Flexible appointment times will be offered to accommodate participants' schedules, reducing barriers to attendance, while appointment reminders will be sent via phone calls to ensure follow-up compliance. Direct contact will also be made available for participants to discuss any concerns or issues they encounter during the study.
Outcome data to be collected if the patient deviates or discontinues
For participants who discontinue or deviate from the trial, the specific reasons for withdrawal, such as adverse events or personal reasons, will be documented. The last available dd-cfDNA levels, LFT results, and Tacrolimus (TAC) levels will be collected, alongside any clinical assessments or adverse events experienced before discontinuation. Follow-up information on the participant's health status post-discontinuation will also be gathered. Updated demographic data will be maintained to assess any potential biases or trends among participants who deviate from the study.
Standardized data entry forms will ensure uniform data collection across participants. A double data entry process will be used, with researchers entering data both on paper and in Excel for validation and error-checking. Automated checks will detect implausible values, and regular audits will correct errors promptly. Data entry personnel will receive training on procedures, with a feedback system in place for ongoing improvement.
Data security will be ensured through restricted access for authorized personnel only, with encryption protecting data during storage and transmission. Secure servers with firewalls and antivirus software will guard against unauthorized access, and regular backups will enable recovery from system failures. Allocation of unique ID numbers will be done to safeguard participant confidentiality. Physical data copies will be stored in locked locations, and long-term storage plans will be developed for future research and data sharing.
A. For analyzing primary & secondary outcomes
• The ROC curve will be used to find an ideal cutoff for cfDNA in predicting the rejection rate among transplant patients.
• To test the statistical significance of the difference in the proportion of rejection rate between two categorizations of dd-cfDNA, and between standard treatment and dd-cfDNA guided treatment, the Chi-square test will be used. Diagnostic measures such as sensitivity, specificity, PVP, PVN, and accuracy will be calculated.
• Pearson's correlation coefficient will be used to study the relationship between dd-cfDNA and DSA. Also, the statistical significance of the correlation will be tested using the regression t-test.
To test the statistical significance of the difference in the proportion of rejection between the two groups, the Chi-square test will be used.
For secondary outcomes, appropriate models will be applied:
• Multivariable regression analysis (linear for continuous outcomes, logistic for binary outcomes) to assess the impact of key covariates on the outcomes.
• If repeated measurements are involved, mixed-effects models or repeated-measures ANOVA will be used.
Subgroup analyses will be conducted to explore potential variations in treatment effects across different groups (e.g., age, sex, disease severity). Stratified analysis may also be performed to compare subgroups independently. The adjusted analysis will account for potential confounding variables using multivariable regression models to adjust for pre-specified covariates (such as age, baseline disease status, graft parameters, and comorbidities). Sensitivity analysis may be performed to explore the robustness of the findings under different modeling assumptions.
C. Definition of Analysis Population Relating to Non-adherence (e.g., Intention-to-treat Analysis) and Methods to Handle Missing Data
The primary analysis will follow the intention-to-treat (ITT) principle, analyzing participants based on their original randomization group, regardless of intervention adherence. A per-protocol analysis may also be performed as a secondary analysis for those who adhered to the protocol. Multiple imputation will be used to handle missing data, assuming it is missing at random (MAR), by generating and analyzing multiple complete datasets and then pooling the results. Sensitivity analyses may assess the impact of different assumptions about missing data, such as missing not at random (MNAR).
A. Components of the Data Monitoring Committee (DMC)
• Summary of the Reporting Committee: The Data Monitoring Committee (DMC) will be composed of independent members with relevant expertise in clinical research, bio-statistics, molecular biology, clinical pharmacy, and transplantation medicine. The primary role of the DMC is to monitor the safety and efficacy data throughout the trial and ensure that the study is conducted ethically and in the best interest of the participants.
• Independence from Sponsor: The DMC will operate independently of the study sponsor. The members of the committee will not have any financial or professional ties to the sponsor that could lead to potential conflicts of interest.
• Competing Interests: A formal statement will be included from all DMC members, declaring any potential competing interests, such as involvement in related research or advisory roles for companies that might benefit from the study outcomes. Any conflicts of interest identified will be managed according to the ethical guidelines for clinical research to ensure transparency and integrity in decision-making.
Interim analyses will occur every 6 months to assess safety and early efficacy. Only the DMC will access these results, keeping researchers, sponsors, and participants blinded to maintain study integrity. The DMC can recommend study modifications or early termination if interim data show harm, overwhelming efficacy, or futility. The final decision to terminate will be made jointly by the DMC, principal investigators, and ethics board based on the interim findings.
Adverse events are not expected in this trial, as the study involves only the modulation of immunosuppression based on cfDNA analysis, which is validated with LFTs and liver biopsies. Since no new drugs or invasive interventions are introduced, the risk of unintended side effects is minimal. However, any serious adverse events (SAEs) related to routine clinical care will still be recorded and reported per ethics committee and regulatory guidelines to ensure patient safety.
Auditing procedures, conducted every 6 months will include a review of the trial protocol, regulatory submissions, and guidelines, along with document checks to ensure compliance with protocols, CRFs, and informed consent forms. Data will be verified to match source documents, and participants' rights and consent will be checked for adherence. Safety monitoring will involve reviewing the management of adverse events (AEs) and serious adverse events (SAEs), while data management systems will be inspected for confidentiality and integrity. Findings will be reported with recommendations, and corrective actions will be taken for non-compliance, followed by additional audits if necessary. Independent auditors from the institutional ethics committee will oversee these audits to ensure objectivity.
Plans to seek IEC approval: The study was approved by the Institutional Ethics Committee (IEC) of Amrita Institute of Medical Sciences [IEC-AIMS-2024-PHARMA-304] on 26.11.2024
Institutional Ethics Committee statement:
The Ethics Committee reviewed the documents pertaining to the study protocol titled ‘Donor-derived circulating cell-free DNA as a rejection biomarker and modulator of immunosuppression following liver transplantation: A randomized controlled trial’ presented by Dr.Lakshmi.V.U as investigator with Dr. Dinesh Balakrishnan, Dept of GI surgery and Dr.Narmadha.M.P, School of Pharmacy as the guides.
After reviewing the protocol and hearing the Investigator, the Committee finds that the study is a randomized controlled study with the primary objective to assess the efficacy of donor-derived cell-free DNA (dd-cfDNA) modulated immunosuppression in reducing acute T-cell mediated rejection following liver transplantation. Additionally, the study aims to evaluate and compare the effectiveness of dd-cfDNA as an early biomarker for detecting allograft rejection in liver transplant recipients, in contrast to conventional liver function tests.
PI submits that dd-cfDNA analysis has not been studied in the Indian population with a significant sample from a high-volume transplant center in a tertiary care setting, especially in live donor liver transplant. Internal Scientific Review Report says that the study benefits early detection of allograft rejection, prompt identification of steroid reftactory rejection and tailoring of immunosuppression. The study is an academic study in public interest. No ethical issues are noted. Approval granted. PI to ensure that the data collected are anonymized and confidential. Data privacy of the subjects shall be protected. It is mandatory that PI shall submit interim report at 6-months intervals about the progress of the study and a detailed final report upon closure of the study to the Ethics Committee.
Any important protocol modifications, such as changes in eligibility criteria, study centers, or outcome analysis, will be promptly communicated to all relevant parties. This includes notifying investigators, the Research Ethics Committee (REC)/Institutional Review Board (IRB), trial registries, regulatory authorities, and journals. Updated protocols will be submitted for approval, and participants will be re-consented if necessary. All changes will be documented and publicly reported as required by trial registries and journals.
A. Who will obtain consent from the trial participant
The primary researcher, who is well-versed in the study protocol, will obtain a signed informed consent form from each trial participant. This consent form, approved by the Institutional Ethics Committee (IEC), will be provided along with detailed information about the study, including its risks and benefits. Participants will also have the opportunity to ask questions and clarify any concerns before signing the form.
The statement of consent from the participant is as follows:
“I have understood all the information provided to me regarding the study. I have had the opportunity to ask questions and I understand the risks and benefits associated with my participation. I acknowledge that my clinical and histological medical data as well as blood samples will be collected for research purposes. I understand that I can withdraw from the study at any time without needing to provide a reason or incurring any costs. I hereby consent to participate in the study.”
B. Additional consent for collection & use of participant data & biological specimen
Additional consent will be sought for the collection and use of participant data and biological specimens for future research. Participants will be informed about how their data and samples may be used, stored, and shared, and will be given the option to consent to or decline this aspect of the study.
Personal data will be collected in a secure manner using unique participant IDs to ensure confidentiality. Identifiable information will be stored separately from clinical and biological data, with access restricted to authorized personnel only to maintain confidentiality throughout the trial and post-trial. After the trial, data will be de-identified for analysis, and long-term storage will comply with data protection regulations.
The authors declare that there are no financial conflicts of interest associated with this study.
While investigators can access necessary data for analysis, restrictions on sensitive or proprietary information will be clearly defined. The agreement will outline the timeline for data access and obligations for sharing with sponsors and regulatory authorities, ensuring the confidentiality and integrity of the trial data.
No direct interventions are involved, so adverse effects are not expected. Follow-up assessments will monitor health outcomes and address any long-term effects. Participants will not be charged for dd-cfDNA analysis, while other analyses are part of standard liver transplant recipient monitoring.
A. Plans for the investigator to communicate trial results to participants, healthcare professionals, public & relevant groups
The dd-cfDNA analysis results will not be shared with participants as the impact and reliability remain unknown till the final analysis. Participants will receive a summary of the trial findings after publication in a peer-reviewed journal. Results will also be shared with healthcare professionals and the public through publications, conferences, and professional networks.
B. Authorship eligibility guidelines & use of professional writers
To qualify for authorship, individuals must make substantial contributions to the study's conception, design, execution, or interpretation; participate in drafting or critically revising the manuscript; approve the final version for publication; and be accountable for all aspects of the work. Professional writers will not be used; all authors will contribute directly to writing to ensure research integrity and reflect their involvement. Any additional support from non-authors will be acknowledged in the final publication.
C. Plans if any for granting public access to full protocol, participant-level data set, and statistical code
Participant-level data and statistical code will be shared in compliance with ethical guidelines and data protection regulations, ensuring appropriate anonymization to protect confidentiality. Access will be granted to qualified researchers and organizations upon request for legitimate scientific purposes while maintaining participant privacy. The timeline for data sharing and the specific repository will be communicated in the study's final publication.
Informed consent10
Plan for collection, lab evaluation & storage of biological specimens in current trial & future use in ancillary studies.
Blood samples (9ml) will be collected from participants at specific time points during the trial and subjected to predefined assays and analytical procedures. These specimens will be securely stored at the Molecular Biology lab, Amrita Centre of Nano Sciences and Molecular Medicine, under optimal temperature conditions and proper labeling. Leftover specimens will be discarded and not used for ancillary studies; if future use is considered, additional consent & samples will be obtained, ensuring both the integrity of the trial and respect for participants' rights.
The study was approved by the Institutional Ethics Committee (IEC) of Amrita Institute of Medical Sciences [IEC-AIMS-2024-PHARMA-304] on 26.11.2024
The primary researcher, who is well-versed in the study protocol, will obtain a signed informed consent form from each trial participant. This consent form, approved by the Institutional Ethics Committee (IEC), will be provided along with detailed information about the study, including its risks and benefits. Participants will also have the opportunity to ask questions and clarify any concerns before signing the form.
OSF Repository: SPIRIT checklist for ‘Donor-derived circulating cell-free DNA as a rejection biomarker and immunosuppression modulation tool in liver transplantation: A randomized controlled trial’.
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
B. Role & responsibility of sponsors- The sponsors will fund the study, ensure regulatory compliance and ethical standards, oversee data integrity, and safety monitoring, and coordinate between investigators to manage resources, without influencing data analysis.
B. Composition, roles & responsibilities of coordinating center, steering committee & end-point adjudication committee, data management team & other members overseeing the study
The coordinating center of the institution included the Dean of the institution, Ethics Committee Members, the Clinical Professors and Transplant surgeons of Gastrointestinal Surgery, the Head of the Department of Clinical Pharmacy, and the Principal of Amrita School of Pharmacy. They will manage trial operations and data integrity. The steering committee will oversee scientific direction and protocol adherence, while the end-point adjudication committee will independently validate outcomes. The data management team will handle secure data collection and confidentiality, with statisticians and safety monitors ensuring compliance with ethical and safety standards.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)