Keywords
deficiency of adenosine deaminase 2, polyarteritis nodosa, vasculitis, mutation, hereditary autoinflammatory diseases, case report
deficiency of adenosine deaminase 2, polyarteritis nodosa, vasculitis, mutation, hereditary autoinflammatory diseases, case report
Deficiency in Adenosine Deaminase 2 (DADA2) is a genetic autosomal recessive disease caused by pathogenic variants of the ADA2 gene called the CERC1 (Cat Eye syndrome Chromosome Region, Candidate 1 gene) on chromosome 22q11.1 Its clinical presentation, similar to that observed in Polyarteritis Nodosa (PAN), could lead to a misdiagnosis, especially when presenting at an early age, and its clinical features vary from vasculitic symptoms to hematological abnormalities and immunodeficiency presentations. PAN is a systemic vasculitis that affects medium vessels and causes multiple symptoms, including cutaneous, renal, cerebral, and arterial manifestations.
We present the cases of two siblings with non-consanguineous healthy parents who were diagnosed with ADA2 deficiency and phenotypic manifestations of PAN. We also performed gene expression screening to identify a previously unreported homozygous variant, in both patients.
We report the case of a 21-year-old Caucasian male with non-consanguineous healthy parents. He had no family history other than that of a sibling with immunoglobulin deficiency. At the age of 5 years, he presented with recurrent fever (lasting 2 weeks), without proof of infection, weight loss, livedo reticularis, or subcutaneous nodules. Extensive infectious studies (including blood, urine, cerebrospinal fluid (CSF), stool cultures, viral serologies, and mycobacterial tests) were negative. Biopsies of the nodule revealed perivascular inflammatory infiltrate with polynuclear neutrophilic cells. A diagnosis of childhood Polyarteritis Nodosa (cPAN) was made (according to the EULAR/PReS classification criteria). The patient had been treated with corticosteroids and azathioprine for 2 years and had been in remission for 5 years. In 2021, he was admitted with headache, vertigo, and ringing in the ears of recent onset. Physical examination showed no abnormalities other than livedo racemosa lesions on the legs. Laboratory test results revealed no abnormalities. Antinuclear Antibodies (ANA), anti-neutrophil cytoplasmic antibodies (ANCA), and immunoglobulin levels were normal. Cerebral magnetic resonance imaging (MRI) showed small cortical foci of bleeding at different ages, with no subarachnoid or intraventricular bleeding. After ruling out all other etiologies of secondary cerebral vasculitis, a flare of PAN was suspected. Additional angiography exploration of the aorta and its branches was performed, which revealed multiple peripancreatic microaneurysms and dividing branches of the hepatic and mesenteric arteries.
Given the rarity of juvenile PAN, another etiology was suspected. After meticulously examining the family medical history, we found interesting information. His 24-year-old brother presented with several infections from an early age, including: rhinopharyngitis, gastroenteritis, chickenpox, and ethmoiditis. He was diagnosed with hypogammaglobulinemia, received monthly intravenous perfusions of immunoglobulin until the age of 7 years. Subsequently, he presented with fever, myalgia, and livedo lesions. A deep nodule biopsy revealed lesions suggestive of PAN. The patient received corticosteroids andazathioprine, which was maintained for 4 years. In 2020, he complained of pain and paresthesia in his feet, and electromyography showed bilateral and symmetrical distal axonal sensitivo-motor damage. The patient was administered steroids and azathioprine.
Since both brothers were diagnosed with PAN, and had similar clinical manifestations, genetic etiology was suspected, notably DADA2, whose clinical manifestations mimic those of cPAN. The DADA2 activity assay is not commonly available; therefore, it could not be performed. Instead, a genetic test was performed. We performed Sanger sequencingto identify a mutation in the CECR1 gene coding for ADA2, in both parents and both brothers. This study revealed a novel ADA2 homozygous pathogenic variant that has, not been previously described in the literature. Both the boys were homozygous for the c.1458–1459insTTGpmutation. (Leu486dup), which was classified as having uncertain significance. The parents were heterozygous. These findings led to the diagnosis of DADA2 in both brothers, which presented as cPAN with fever, livedo, hemorrhagic strokes and abdominal aneurysms with the first, immunoglobulin deficiency and peripheral nervous system involvement with the second. To our knowledge, this mutation has not been identified or reported in the medical literature.
Both patients were treated with Tumor Necrosis Factor inhibitors (TNFi) (Infliximab 3–5 mg/kg per dose) with no documented side effects. There were no relapses during the 2-year follow-up period.
Timeline of clinical features and interventions are summarized in Figure 1.
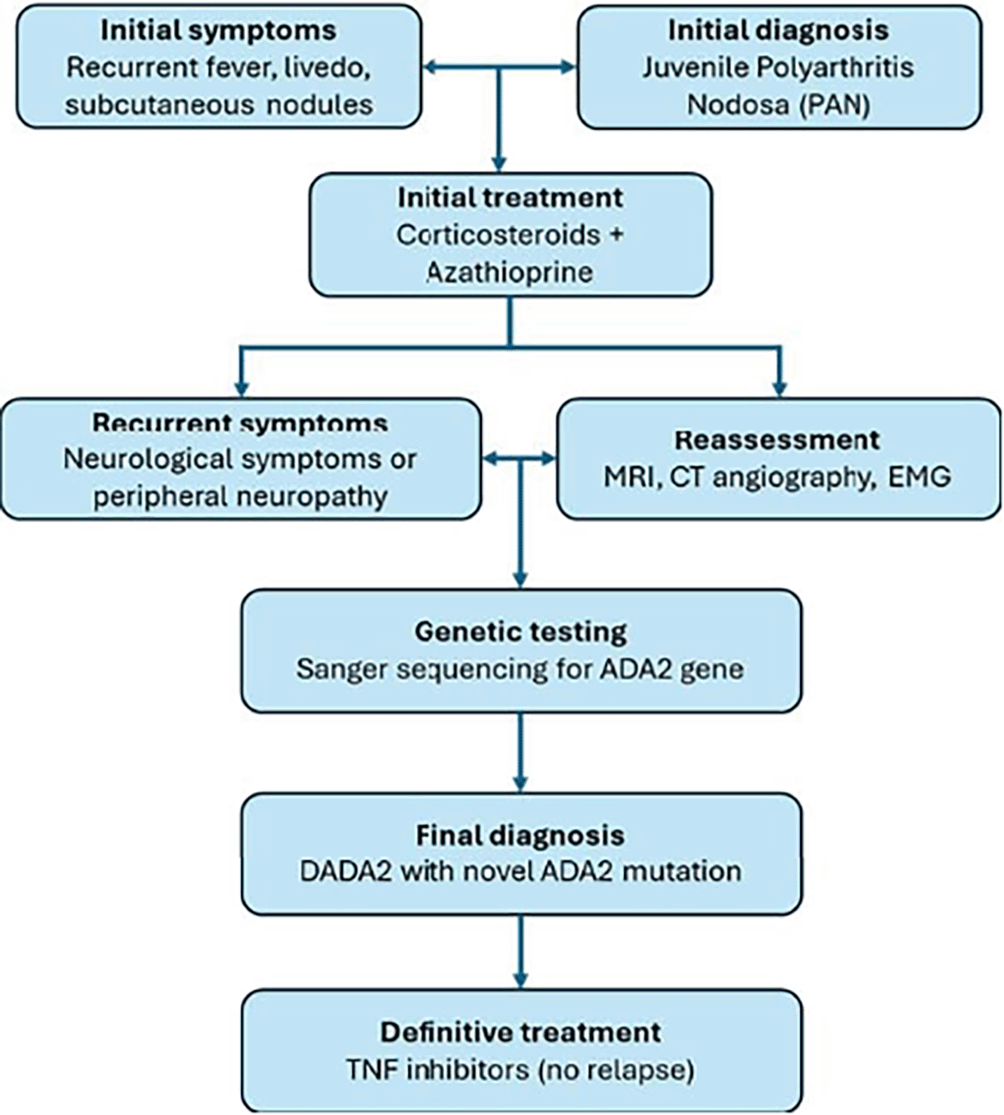
This flowchart summarizes the clinical sequence from initial symptoms and misdiagnosis of juvenile polyarteritis nodosa (PAN) to the final diagnosis of Deficiency of Adenosine Deaminase 2 (DADA2) confirmed by genetic testing.
DADA2 is an autoinflammatory disease caused by a loss-of-function mutation in CECR1, which encodes the ADA2 enzyme. ADA2 is a dimeric enzyme expressed in cells of the myeloid lineage, and monocytes differentiate into macrophages and dendritic cells.2 It acts in extracellular space as an endothelial growth factor in.2
Biopsy samples taken from the brain and skin lesions of patients with DADA2 demonstrated significant endothelial damage and increased staining for interleukin-1β, inducible nitric oxide synthase, and TNFα.1 These findings explain the pro-inflammatory phenotype with endothelial damage and vasculitis.
ADA2 regulates extracellular adenosine levels. Adenosine is known to play a role in the inflammatory responses during acute conditions. ADA2 deficiency causes dysregulation of adenosine levels, leading to chronic inflammation.
NETosis is another mechanism found in DADA2 patients. Indeed, M1 macrophages incubated with NETs from patients with DADA2 release abundant TNFα.
DADA2 phenotypes are associated with mutations inADA2 on chromosome 22q11.1. One hundred fifty-eight mutations were reported in the Infevers database, an online registry of hereditary auto-inflammatory disorder mutations, until March 2025. Of these, 115 mutations were found to be pathogenic/likely pathogenic: 89 substitutions (77, 4%), 21 deletions (18, 3%), four duplications, and one deletion-insertion (del-ins).3 Only one mutation was found in a symptomatic patient of Tunisian origin. This is a substitution for exon 2, c.73G>Tp. (Gly25Cys).
Our patients were homozygous carriers of a previously unreported mutation c.1458–1459insTTGp. (Leu486dup), resulting in different phenotypes, and is classified as of uncertain significance.
Indeed, multiple studies have concluded that there is a large variability in phenotypic manifestations. Differences in phenotypic presentations may occur with the same mutation, including in patients within the same family with the same mutation.4 This suggests the involvement of epigenetic and environmental factors that influence the phenotype and severity of the disease.
Screening for DADA2 is based on two diagnostic methods: measurement of plasma/serum ADA2 activity and genetic testing. To date, no definitive criteria have been established for this condition.
Puietal. suggested an international consensus for screening DADA2.5 The DADA2 Consensus Committee, consisting of patient representatives and global experts representing adult and pediatric subspecialties, developed statements for diagnostic testing and screening of DADA2. The diagnosis can be determined by either measurement of ADA2 activity and/or sequencing of the ADA2 gene. Both methods should be used if available.5 All patients with DADA2 described to date have very low or undetectable ADA2 catalytic activity in plasma or serum. When available, this method should be performed by a certified laboratory, and normal levels of ADA2 activity are sufficient to rule out diagnosis.5
Genetic testing consists of the targeted sequencing of ADA2. DADA2 is diagnosed by the presence of biallelic or likely pathogenic ADA2 variants, as determined by the guidelines established by the American College of Medical Genetics and Genomics.5
It is performed either by conventional Sanger sequencing, next-generation sequencing (NGS)-based targeted panels, or exome sequencing.6 Small deletions and duplications missed by routine techniques may be detected using multiplex ligation-dependent probe amplification (MLPA) and long-range polymerase chain reaction (PCR).
Patients with monoallelic pathogenic or likely pathogenic ADA2variants are considered carriers.
For patients with monoallelic or biallelic ADA2variants of unknown significance (VUS), measurement of ADA2 enzyme activity should be strongly considered to evaluate the possibility of DADA2.5 For Families with a confirmed case of DADA2, screening of siblings should be considered, even in the absence of symptoms, since they are independently at risk and the disease may be, asymptomatic.5
The clinical manifestations of DADA2 have varied widely and expanded since 2014. In addition to the pan-like phenotype, autoinflammation, immunodeficiency, hematological involvement, and lymphoproliferation features have been described.6 There are also asymptomatic or mildly symptomatic cases.6
To date, more than 600 cases of DADA2 have been reported. This disease seems to be more common in Caucasians and South Asians. The age of presentation ranges from at birth to 59 years.2,6 The age of onset is mostly pediatric; however, adult presentation (between 18 and 59 years) has been well documented.6
The first case described a PAN-like phenotype.1 The most common clinical manifestations are cutaneous, neurological, and recurrent fevers. Second, the gastrointestinal, renal, cardiac, and pulmonary systems.6
The most common skin manifestation is livedo reticularis/racemose.2,6 Skin vasculitis can present as subcutaneous nodules (including erythema nodosum), purpuric lesions, Raynaud’s phenomenon, skin ulcers, mouth and genital ulcers, urticarial rash and digital gangrene, as well.7
Neurological involvement is common and may be a form of disease.8 Neurological manifestations may affect both the central nervous system (CNS) and/or the peripheral nervous system (PNC).6,8 Stroke, particularly ischemic stroke, is the most common neurological manifestation.1,8 In a review of neurological manifestations of DADA2, including 628 patients, Dzhusandet al.8 reported that strokes accounted for 77.5% of all neurological events. Other neurological symptoms include mononeuropathies and polyneuropathies; focal neurological deficits including cranial nerve paralysis; ophthalmological findings such as strabismus, optic neuritis, uveitis, loss of vision, seizures, headache; and other a specific symptoms such as vertigo and amnesia.6,8 Approximately3% of the patients reviewed had neurocognitive disorders, including isolated cognitive impairment, isolated memory disturbance, autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), isolated persistent irritability, and isolated difficulties in concentration.8 This is particularly interesting, as neurocognitive disorders may be the sole manifestation of DADA2 and should be examined in a pediatric population in whom cognitive disorders are possibly misdiagnosed.
MRI is the preferred imaging modality, and reveals that ischemic strokes are preferentially localized in the brain stem, thalamus, basal ganglia, internal capsule, and other deep cerebral structures. The Negativity of MRI does not exclude the diagnosis, as there were cases of neurological signs with a mild or transient character (transient ischemic attacks (TIA)), or even cases of MRI-negative infarcts.8 Hemorrhagic strokes were described in 11% of cases, mostly in lobar locations. However, basal ganglia,8 subdural,1 and intraventricular hemorrhage may occasionally occur. They may be complicated by the use of antiplatelet agents and anticoagulants in patients with a history of ischemic stroke.6 Other MRI abnormalities may include brain atrophy, vessel wall enhancement, posterior reversible encephalopathy syndrome (PRES) pattern, optic atrophy, aneurysms, arterial stenosis, and microhemorrhage.8
Renal involvement presents as hypertension, renal infarcts, kidney artery aneurysms and glomerulonephritis.9 Testicular involvement, including pain or swelling, is reported to be one of the features similar to PAN. Gastrointestinal involvement mimics the gastrointestinal features of PAN. Abdominal pain can be a unique symptom of mesenteric ischemia and may lead to bowel perforation if it is not diagnosed at time.9 Stenosis and aneurysms of the celiac and mesenteric arteries, as described in the first patient, have been reported.10 Cardiac involvement appears as dilated cardiomyopathy, coronary aneurysms, and focal myocarditis.9
Most patients with a vasculitic phenotype fulfill the criteria for PAN, including angiographic and histopathological features.11 However, patients with DADA2 are relatively younger than those with PAN and are more likely to have renal and neurological involvement, especially strokes. Other DADA2 phenotypes include Sneddon’s syndrome, antiphospholipid antibody syndrome, and Behçet’s disease.8
Hematological involvement is well-documented and may occur solely or in combination with other DADA2 phenotypes. Anemia is common and results from erythroid hypofunction. It presents as moderate anemia of nutritional etiology, autoimmune hemolytic anemia, or severe refractory anemia similar to pure red cell aplasia (PRCA) and Diamond Blackfan anemia (DBA).12
Other hematological cytopenia include leukopenia, neutropenia, thrombocytopenia, and pancytopenia. Autoimmune cytopenia, including Coombs positive test with or without hemolytic anemia and immune thrombocytopenia,6 and features of hemophagocytic lymphohistiocytosis have also been reported.1
Recurrent non-infectious fever is common. Other symptoms suggestive of autoinflammatory syndrome include recurrent episodes of arthralgia/arthritis and myalgias/myositis. Positive Anti-nuclear antibodies and few cases with anti-neutrophil cytoplasmic antibodies (ANCA) and antiphospholipid antibodies were noted, without a clear significance yet.
DADA2 is associated with a mild immunodeficiency including humoral immunodeficiency with low immunoglobulin levels (particularly low IgM levels) as the most prevalent abnormality.6,7,9 Additionally, hypogammaglobulinemia, as described in the second patient, impaired vaccine responses, B cell lymphopenia, and low-switched memory B cells are increasingly being reported. The immunodeficiency phenotype can be misdiagnosed as Common Variable Immunodeficiency (CVID), and the presence of vasculitic features may lead to DADA2 diagnosis.12
Splenomegaly with or without hepatomegaly as well as lymphadenopathy are well reported features of DADA2.7,12 Lymphoproliferation in DADA2 includes T-large granular lymphocytic leukemia (T-LGLL), autoimmune lymphoproliferative syndrome (ALPS)-like disease, and Hodgkin Lymphoma.9 A multicentric Castleman’s disease like phenotype was reported as well.12
Lee et al. elaborated consensus statements for the management of DADA2 based on published case series and experts’ opinions.5
Unlike in patients with PAN, the efficacy of glucocorticoids in the management of DADA2 flares seems to be limited. Indeed, most patients show a partial response to high doses of glucocorticoids with relapses on tapering or are refractory to steroids.5,6 Retrospective studies have demonstrated the beneficial role of tumor necrosis factor inhibitors (TNFi) in controlling the inflammatory and/or vasculitic flares of the disease. TNFi have been proven to lower the risk of ischemic and hemorrhagic stroke.5 The use of disease-modifying antirheumatic drugs (DMARDs), such as methotrexate, azathioprine, cyclophosphamide, cyclosporine, and tacrolimus, has been described, but their long-term efficacy remains limited. Other biological agents have been used, including biologics targeting interleukin 1 (IL1) and 6 (IL6), with no proven or sustainable efficacy.5
Disease flares may occur under TNFi treatment in cases of suboptimal dosing of TNFi, treatment discontinuation, or the development of anti-drug antibodies, such as anti-drug antibodies to infliximab, adalimumab, and golimumab. The use of methotrexate may prevent neutralizing anti-drug antibodies and regulate serum TNFi levels.
Allogenic hematopoietic stem cell transplantation (HSCT) is a proven curative option, especially for patients with hematological manifestations and avasculitic phenotype refractory to immunomodulators. Intravenous immunoglobulin (IVIG) cures are considered in patients with humoral immunodeficiency and recurrent infections.
Our patients were both treated with TNF inhibitors, with no relapse noted during a 2-year follow-up.
Identification of a novel ADA2 variant in these siblings broadens the mutational spectrum of DADA2 and highlights the genetic heterogeneity of the disease. Given the phenotypic overlap with early onset polyarteritis nodosa, misdiagnosis remains a significant risk, potentially delaying targeted therapies, such as anti-TNF agents. While whole exome sequencing enabled the diagnosis in this case, the lack of enzymatic ADA2 activity assays is a limitation, as functional validation is essential to confirm pathogenicity. Further studies are needed to elucidate the impact of specific mutations on ADA2 function and clinical phenotype, and to define standardized diagnostic workflows in resource-limited settings. The identification of this novel mutation may reflect ethnic, genetic, epigenetic, or environmental differences specific to our region. It is plausible that this variant is unique to the Tunisian population. Further studies are needed to explore and validate this hypothesis.
DADA2 is a genetic autoinflammatory disease with clinical, biological, and imaging features similar to those of PAN with childhood onset. The distinction between these diseases can be challenging but is crucial because their treatments are different. We report the cases of two siblings with different phenotypes of DADA2 who were initially misdiagnosed with PAN. Indeed, it is fundamental in the diagnostic approach to investigate the family history of a young patient presenting with multiple symptoms suggestive of vasculitis. Treatment options for the disease are limited, and the outcomes of biologic agents, although efficient, remain unclear. More studies are needed to elucidate the genetic and physiopathologic mechanisms, as well as the role of epigenetic and environmental factors in the development and severity of the disease. Further research is needed to find a targeted therapy that can cure the disease without harmful side effects.
Feedback from the patients and caregivers was actively sought. Both patients and their parents reported a significant improvement in quality of life after treatment, with better disease control. They expressed high levels of satisfaction, particularly with their ability to attend school regularly, which in turn improved their social interactions and general well-being.
MRI | Magnetic Resonance Imaging |
CT | Computed Tomography |
EMG | Electromyography |
ADA2 | Adenosine Deaminase 2 |
DADA2 | Deficiency of Adenosine Deaminase 2 |
TNF | Tumor Necrosis Factor |
Written informed consent for publication of their clinical details was obtained from the patients.
Extended data, including the CARE checklist, consent form template and a figure are available on Zenodo, doi.org/10.5281/zenodo.16746865
I. Ben Hassine: [email protected]
S. Naija: [email protected]
W. Baya: [email protected]
N. Adaily: [email protected]
M. Karmani: [email protected]
J. Anoun: [email protected]
H. Boudriga: [email protected]
A. Mzabi: [email protected]
A. Rezgui: [email protected]
F. Ben Fredj: [email protected]
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)