Keywords
microbiopsy; minimally invasive skin sampling; experimental dermatology; molecular dermatology; transcriptomics; precision diagnostics; skin biomarkers
This article is included in the Bioinformatics gateway.
microbiopsy; minimally invasive skin sampling; experimental dermatology; molecular dermatology; transcriptomics; precision diagnostics; skin biomarkers
Minimally invasive skin sampling has become an increasingly important part of experimental and translational dermatology because many modern questions require molecular, cellular, microbial, or functional readouts rather than histology alone. A growing set of approaches can now collect skin-associated material with less burden than conventional biopsy, including swabs, tape strips, hair plucking, suction blistering, microneedle patches, skin scraping, absorbent microsampling, and microbiopsy.11 These methods differ substantially in the layer sampled, analytes recovered, spatial precision, participant burden, degree of procedural perturbation, and suitability for repeat sampling.
This creates a practical problem for researchers: the least invasive method is not always the most informative, and the most informative method is not always feasible for serial, paediatric, cosmetically sensitive, or field-based studies. Surface approaches may be ideal for broad stratum-corneum, microbiome, or barrier-associated questions, whereas questions involving viable cells, sub-surface infection, lesion microheterogeneity, treatment-response biology, or paired molecular and functional assays may require a method that reaches deeper while still avoiding the morbidity of a conventional biopsy. The rationale for this review is therefore not simply that conventional biopsy is invasive, but that the expanding menu of minimally invasive technologies makes fit-for-purpose method selection increasingly important.
Conventional punch and shave biopsies remain essential when tissue architecture and histopathologic diagnosis are required. However, they remove millimetres of tissue, typically require local anaesthesia, and may leave a wound, which limits repeated sampling, spatial mapping, paediatric studies, and work in cosmetically sensitive sites.1,2 Throughout this review, microbiopsy refers to the sub-millimetre skin-sampling platform originally engineered for minimally invasive tissue recovery; the term micro-punch biopsy (MPB) is used only where individual studies used that nomenclature for closely related devices. Microbiopsy was conceived as a middle-ground technology: more informative than purely surface-based sampling, yet substantially less invasive than a 2–4 mm punch biopsy.1,2 The platform is designed to collect viable cells from the epidermis to the papillary dermis, enabling nucleic acid-based or functional analyses while reducing tissue burden.2,4,5
Within the broader landscape, Hadeler et al. surveyed 96 articles spanning 13 minimally invasive biosampling methods and positioned microbiopsy between surface-based tools such as tape stripping or swabbing and more disruptive procedures such as punch biopsy or suction blistering.11 In experimental terms, tape stripping primarily informs barrier and terminal-differentiation processes, suction blistering facilitates immune-centric analyses at the cost of procedural perturbation, and punch biopsy enables architectural resolution but limits repeat sampling. Microbiopsy occupies an intermediate niche, trading spatial context and tissue architecture for longitudinal molecular accessibility. This review builds on that perspective by examining microbiopsy in detail: how the technology evolved, which research questions it has already supported, which analytical workflows have succeeded or failed, and where microbiopsy should be considered complementary rather than universally superior to other minimally invasive methods.
This framing also aligns microbiopsy with hypothesis-driven skin biology rather than diagnostics alone. The method is most informative when paired with clearly specified experimental questions, such as how local barrier disruption alters viable-cell responses, how lesion-edge molecular signals evolve over time, or how topical perturbation models can be sampled repeatedly without the burden of serial punch biopsies.
The original skin microbiopsy device described by Lin and colleagues was fabricated from three stacked stainless steel plates tapered to a point, with a central chamber designed to mechanically retain a minute tissue specimen2 ( Figure 1). in vivo reflectance confocal microscopy showed a puncture site width of approximately 0.21 ± 0.04 mm, while histology in excised skin demonstrated puncture sites 0.22 ± 0.12 mm wide and 0.26 ± 0.09 mm deep.2 Wound closure occurred rapidly: erythema resolved within 24 hours, the site was not visible to the naked eye after 7 days, and confocal evidence of a small crust disappeared by 3 weeks.2 These data established microbiopsy as a genuinely low-burden sampling approach rather than simply a miniaturized biopsy.
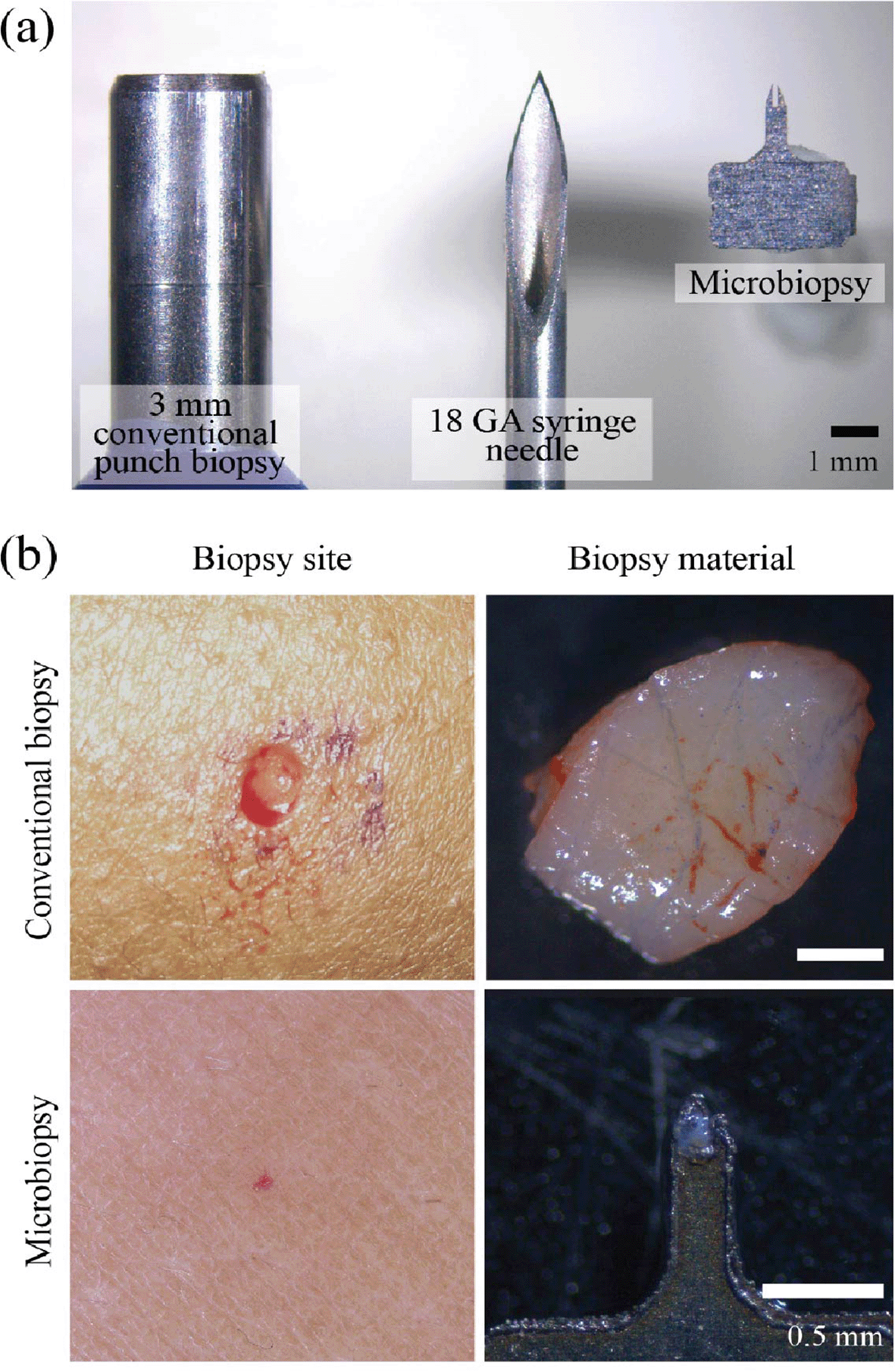
The same engineering study also demonstrated that yield was highly sensitive to device geometry and deployment mechanics. A 0.15 mm channel width achieved the highest average DNA recovery (5.9 ± 3.4 ng), whereas application velocities below 9.2 m/s produced negligible DNA and a velocity of 16.6 m/s markedly improved collection efficiency.2 Despite the very small specimen size, optimized sampling yielded both DNA and RNA (9.0 ± 10.1 ng RNA on average), and downstream amplification workflows supported proof-of-concept transcript analysis.2 These optimization studies were essential because they showed that microbiopsy performance depends not only on the device footprint, but also on roughness, channel geometry, and applicator dynamics.
A second major methodological step was the development of the absorbent microbiopsy, which inserted an absorbent layer between steel plates to collect skin and blood simultaneously.7 This modification broadened the analyte space without substantially increasing clinical burden. In the JoVE protocol study, a 10-second post-application hold improved average RNA recovery from 0.33 ± 0.39 ng to 1.43 ± 0.88 ng and increased blood capture while maintaining skin-marker recovery.7 In practical terms, this evolution transformed microbiopsy from a pure tissue-harvesting microneedle into a hybrid microsampling platform with immediate translational relevance for inflammatory and vascularized skin conditions.
Published studies demonstrate that microbiopsy is adaptable to a wide range of sampling contexts. Wart studies sampled the palm, thumb, finger, and perilesional skin at measured distances from the lesion.4 Oncologic case reports sampled pigmented lesions on the mid-back and peri-lesional skin for internal comparison.5 Device-development and absorbent microbiopsy studies used healthy forearm or volar arm skin for optimization and biomarker profiling.2,7,8 In the Munich Atopy Prediction Study (MAPS) birth-cohort protocol, microbiopsies are planned for lesional and non-lesional atopic dermatitis skin, with matched controls, thereby extending the method into paediatric longitudinal research.9
Feasibility outcomes are similarly encouraging. The original device generated low pain scores, with an average of 1.5 ± 1.1 in volunteers under optimized conditions, and all volunteers scored pain as 0 five minutes after the final application.2 The absorbent version produced only minor erythema and application sites that were not noticeable after 48 hours.7 In the HPV study, microbiopsy achieved comparable viral detection to swabbing but with greater positional precision because the puncture footprint was roughly 0.2 × 0.1 mm, compared with approximately 3 mm for a standard punch biopsy.4 Collectively, these observations support microbiopsy as a feasible option when the key requirement is precise, repeated, or cosmetically conservative molecular sampling rather than full architectural pathology.
More recent work broadened feasibility beyond classic dermatology lesions. In Ethiopian and Indian leishmaniasis studies, microbiopsy was used on papulo-nodular, plaque, crusted, and mucocutaneous lesions as well as on clinically normal skin at the arm and neck to quantify parasite burden across disease states.12,13 In a veterinary proof-of-concept study, pooled microbiopsies could be collected from awake client-owned dogs at inguinal or axillary sites, again with good tolerability in routine clinical handling.16 These reports suggest that microbiopsy is adaptable not only across anatomical sites but also across infectious-disease and comparative-medicine settings.
Fragile-skin cohorts add a distinct feasibility question. Author-provided, non-peer-reviewed EBS pilot material suggests that microbiopsy may be acceptable in fragile skin contexts where conventional diagnostic biopsy can be difficult or refused, and that targeted keratin expression can be explored from pooled samples.18 This evidence should be treated as provisional perspective material rather than as a basis for performance claims until peer-reviewed publication status is confirmed.
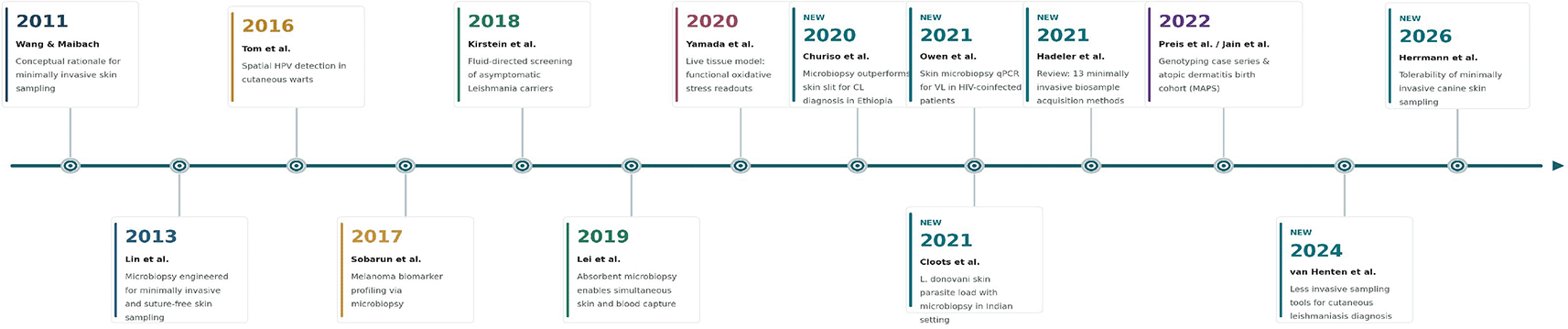
Figure 2 provides a chronological overview of representative studies that shaped the microbiopsy field. The progression from early device development to clinical diagnostics, functional testing, cohort-based research, and comparative benchmarking highlights how microbiopsy has moved from proof-of-concept sampling toward broader translational applications. Teal-bordered entries indicate papers newly incorporated into the present review.
One of the earliest clinical applications focused on HPV DNA detection in cutaneous warts. Tom and colleagues sampled two patients using both microbiopsy and swabbing, then performed type-specific PCR and sequencing for wart-associated HPV genotypes4 ( Figure 3). Microbiopsy DNA yield was only 1–2 ng, yet HPV27 and HPV4 could still be detected accurately in lesional material.4 Importantly, in one case swabbing detected HPV DNA in perilesional skin 1 cm from the wart while microbiopsy did not, a difference that likely reflected the much larger area sampled by the swab.4 This study highlighted a recurring microbiopsy advantage: lower sample mass may be a limitation for broad screening, but it improves spatial specificity when the clinical question concerns lesion margins or microheterogeneity.
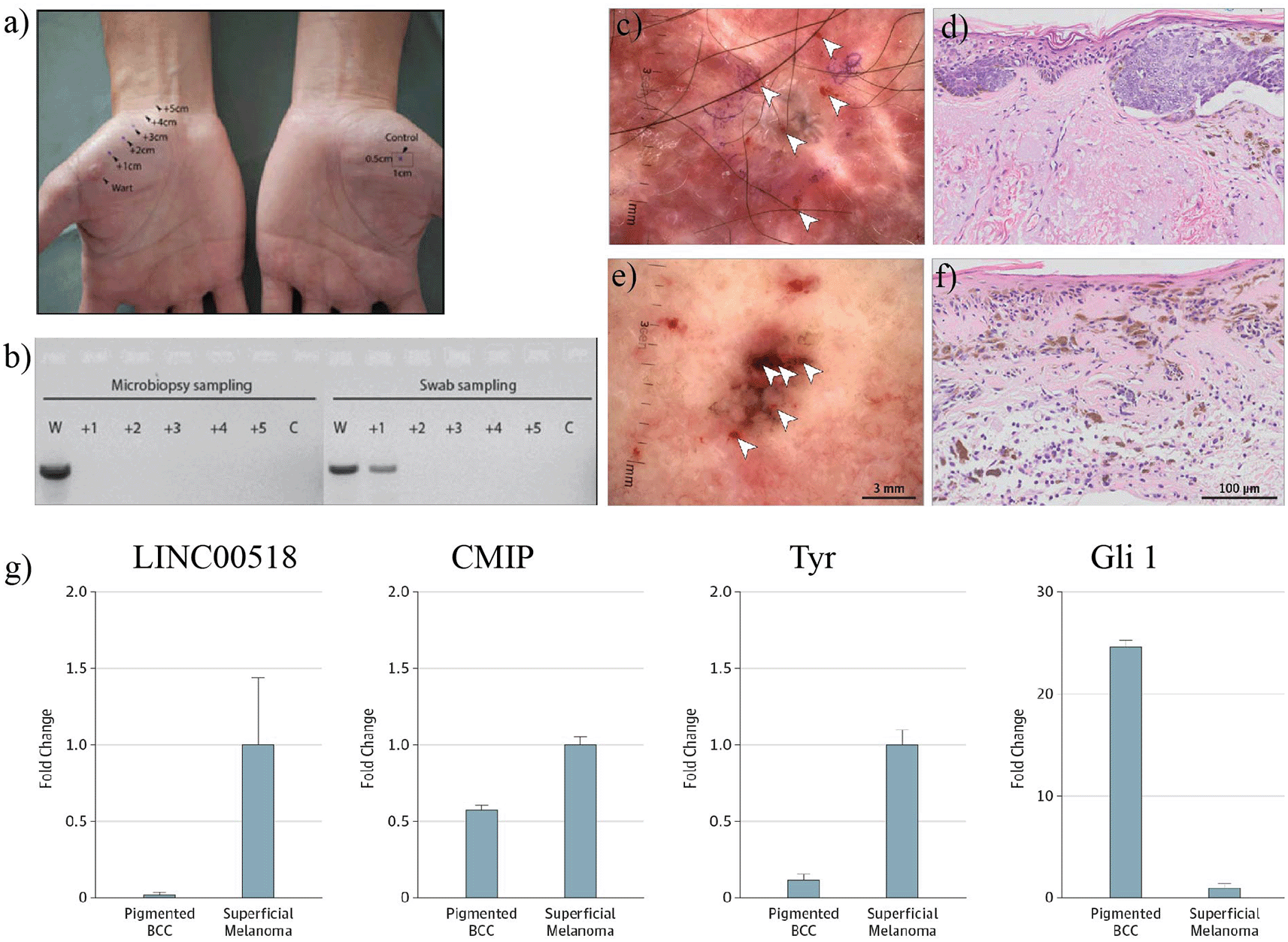
(Top panels)- HPV wart and perilesional sampling layout with PCR detection; (Bottom panels)-Dermoscopic/histologic and qPCR biomarker comparison for melanoma versus pigmented BCC Source panels adapted from Tom et al.,4 Sobarun et al.,5 and Jain et al.10
Microbiopsy has also been used to discriminate diagnostically challenging pigmented lesions ( Figure 3). In a case report comparing a superficial melanoma with a pigmented basal cell carcinoma, three pooled microbiopsies per lesion were analyzed by qPCR for TYR, LINC00518, CMIP, and GLI1.5 LINC00518 was upregulated 47-fold in the melanoma sample, TYR expression was 10-fold higher than in the BCC comparator, and GLI1 was 24-fold higher in the BCC control.5 Histopathology subsequently confirmed lentiginous melanoma in the index lesion, demonstrating that microbiopsy-derived biomarker profiles can provide actionable molecular discrimination even when dermoscopic morphology is equivocal.5
The most direct move toward genomic triage was the 2022 case series by Jain et al., which evaluated micro-punch biopsy devices for BRAF V600E testing in melanocytic lesions.10 Five adults with six lesions underwent lesion and peri-lesional sampling with both skin and blood-format MPB devices, followed by whole-genome amplification and digital droplet PCR.10 Average DNA recovery after amplification was 21 μg, mutation status agreed 100% with reference immunohistochemistry, and there was no significant difference in DNA yield between the two device formats.10 Although the cohort was small, the study provided proof that microbiopsy can support clinically relevant genotyping workflows rather than merely exploratory biomarker discovery.
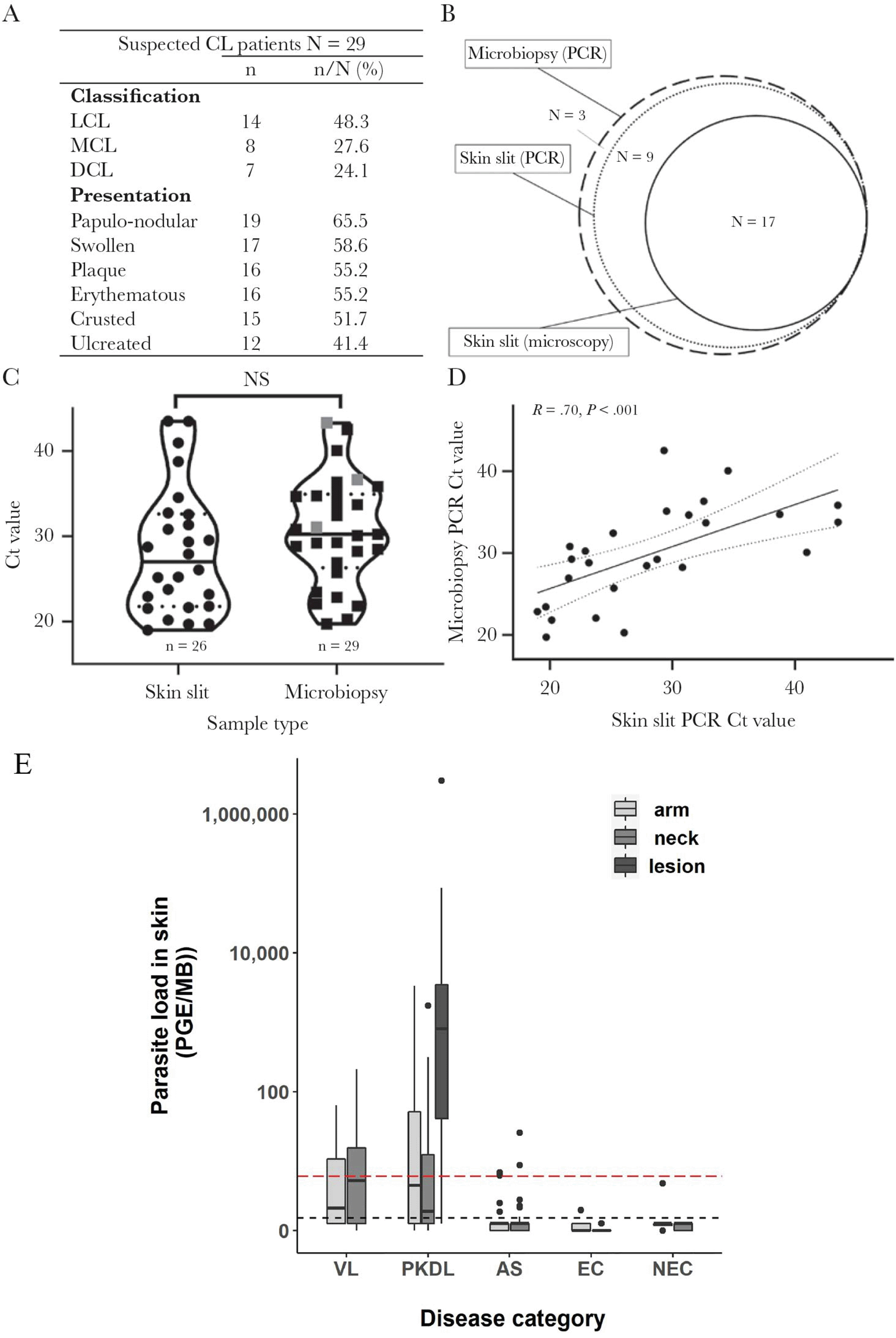
Parasitology has provided some of the clearest demonstrations of microbiopsy’s unique diagnostic niche. In Ethiopian cutaneous leishmaniasis, Churiso et al. reported that microbiopsy PCR confirmed all 29 clinically suspected cases, whereas skin-slit PCR confirmed 26 of 29, and the self-reported pain score was substantially lower for microbiopsy than for skin-slit sampling.12 Because many lesions were non-ulcerated or dry, this study highlighted the practical advantage of a device that can sample below the surface without requiring a conventional incision.12
The concept was extended in India, where Cloots et al. used microbiopsy qPCR to compare skin parasite load across visceral leishmaniasis (VL), post-kala-azar dermal leishmaniasis (PKDL), asymptomatic infection, endemic controls, and non-endemic controls.13 At least one skin sample was positive in 55% of VL patients and 86% of PKDL patients, four skin samples from three asymptomatic individuals were positive, and PKDL lesion-edge samples carried much higher parasite burdens than arm or neck skin.13 These findings repositioned microbiopsy from a point diagnostic tool to a method for studying transmission biology and clinically relevant cutaneous reservoirs.
Microbiopsy is not limited to diagnostic sampling; it can also support functional and toxicologic experimentation in living tissue. Yamada et al. used microbiopsy plus confocal live-cell assays to assess oxidative-stress readouts after topical exposure to zinc oxide nanoparticle sunscreen in six volunteers8 ( Figure 4). Tape stripping increased transepidermal water loss, confirming barrier disruption, and tert-butyl hydroperoxide positive controls generated the expected increases in reactive oxygen/nitrogen species and mitochondrial superoxide signals.8 In contrast, zinc oxide nanoparticle treatment did not produce a significant increase in oxidative-stress markers in intact or tape-stripped human skin under the conditions tested.8 The importance of this study was methodological as much as biological: it showed that microbiopsy can recover viable tissue suitable for immediate ex vivo functional readouts rather than only endpoint nucleic-acid analysis.
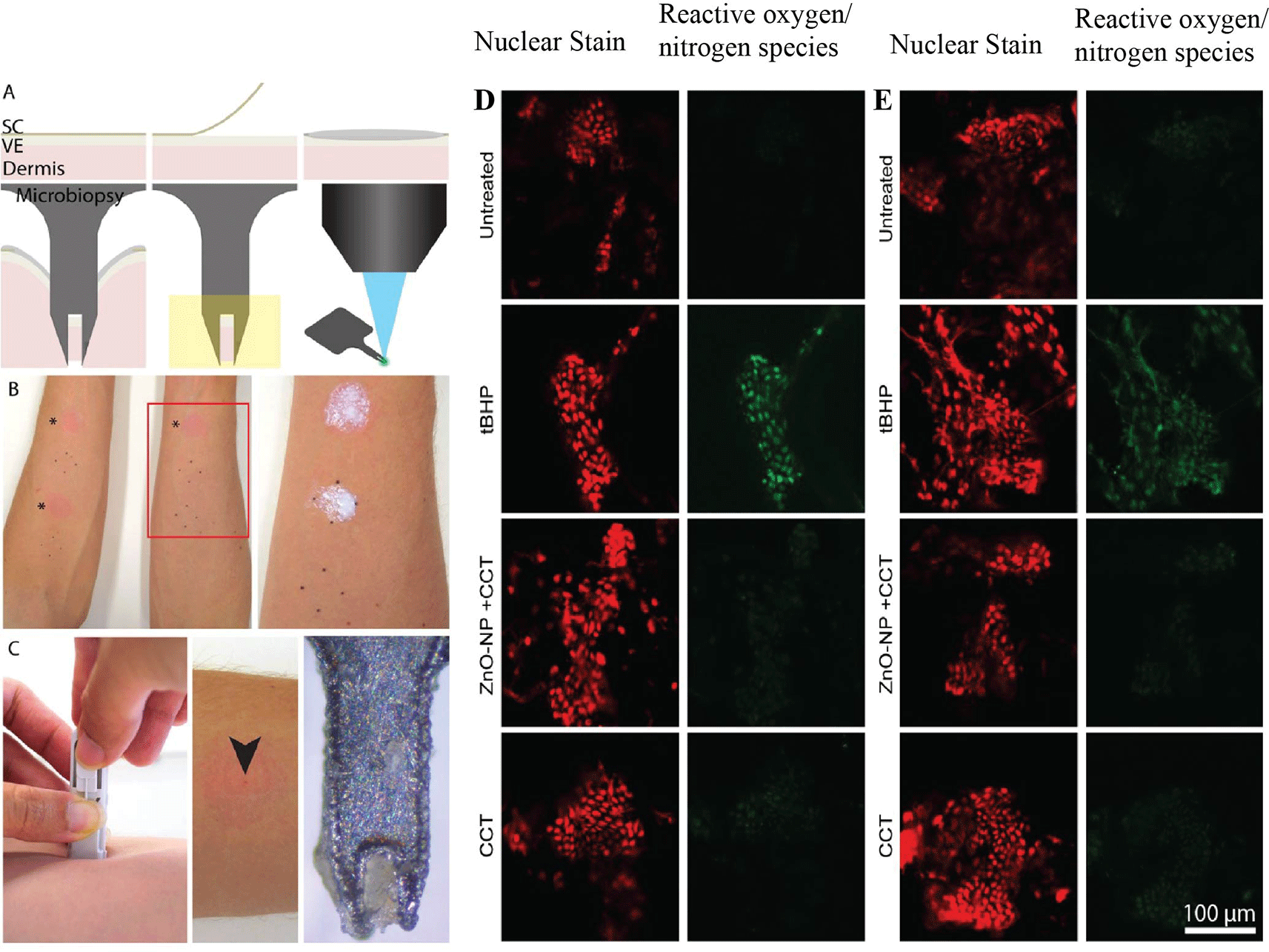
The absorbent microbiopsy protocol extended this logic by recovering both skin and blood-associated markers from a single application. Relative RT-qPCR signals for TYR and KRT14 were comparable between skin and absorbent microbiopsies, whereas the absorbent device yielded stronger CD3E and CD19 signals, consistent with enhanced blood capture.7 This dual-compartment recovery suggests a future role in treatment-response studies where epidermal biomarkers, infiltrating immune cells, and blood contamination all carry biological meaning. In other words, microbiopsy can sample not just structure, but process.
The low visible morbidity of microbiopsy makes it well suited to serial or longitudinal designs. Even small proof-of-concept studies already used spatial repetition within the same participant, such as microbiopsy and swab comparisons at defined distances from warts or matched lesion-versus-perilesion sampling in pigmented tumors.4,5 The more ambitious example is the MAPS protocol, which embeds microbiopsies into a 300-child birth cohort followed from early infancy to 48 months of age.9 In that study design, microbiopsy-derived transcriptomics is interpreted alongside skin-barrier measures, microbiome sequencing from skin and stool, prick testing, blood phenotyping, and detailed environmental exposure data.9
This longitudinal framing is important because it shifts microbiopsy from a single-site laboratory method to an enabling platform for disease-course modelling. In atopic dermatitis, for example, serial lesional and non-lesional transcriptomes may be aligned with clinical severity, barrier dysfunction, and microbiome composition over time.9 Similar logic could be applied to wound healing, inflammatory dermatoses, topical drug studies, or lesion evolution in pigmented neoplasia. Repeated conventional biopsies are often unacceptable in these settings; repeated microbiopsies are considerably more plausible.
The planned VL-HIV study by Owen et al. provides a second model for longitudinal deployment. In that design, skin microbiopsy qPCR is scheduled alongside blood, urine, and parasitological reference tests at day 0 and repeated at days 3, 8, 15, and 29 to assess both diagnosis and test of cure.14 If such workflows are validated, they would demonstrate one of microbiopsy’s strongest practical advantages: the ability to revisit the same biological question over time without the burden of repeated splenic, bone marrow, or conventional cutaneous biopsy.
Table 1 summarizes the evolution of skin microbiopsy from early minimally invasive sampling concepts to current applications in dermatology, infectious disease, cohort-based research, and comparative medicine. The timeline highlights how the field has moved beyond device development toward practical research questions, including spatial viral detection, lesion biomarker profiling, experimental perturbation testing, parasite-load assessment, longitudinal transcriptomics, and cross-species feasibility studies. This progression supports the rationale for reviewing microbiopsy not as a single-purpose biopsy replacement, but as one of several emerging minimally invasive tools whose value depends on the biological question, target analyte, sampling depth, downstream assay, and tolerance for loss of tissue architecture.
| Study | Device/cohort | Sampling context | Readout | Main contribution |
|---|---|---|---|---|
| Lin 20132 | Skin microbiopsy; 20 healthy volunteers plus ex vivo AK work | Healthy skin and matched excised lesions | DNA/RNA yield, RCM, dermoscopy, wound healing, pain | Established the sub-millimetre platform and identified key engineering parameters for reliable molecular sampling. |
| Tom 20164 | Skin microbiopsy; 2 patients | Warts and perilesional skin on palm/finger | HPV PCR and sequencing | Demonstrated spatially precise viral DNA detection with very small footprints. |
| Sobarun 20175 | Skin microbiopsy; case report | Pigmented back lesions and perilesional skin | qPCR biomarkers (TYR, LINC00518, CMIP, GLI1) | Showed molecular discrimination of melanoma from pigmented BCC. |
| Lei 20197 | Absorbent microbiopsy; volunteer protocol | Volar arm | RNA extraction and RT-qPCR for skin and blood markers | Added simultaneous skin and blood capture; highlighted dwell-time effects on yield. |
| Yamada 20208 | Skin microbiopsy; 6 volunteers | Inner forearm, intact and tape-stripped skin | Confocal live-cell oxidative-stress assays | Provided a minimally invasive human model for treatment/toxicity testing in viable tissue. |
| Jain 202210 | Skin- and blood-format MPB; 5 adults/6 lesions | Lesional and peri-lesional melanocytic lesions | DNA amplification, ddPCR, IHC comparison | Supported BRAF V600E genotyping with full concordance to reference testing. |
| MAPS 20229 | Prospective birth-cohort protocol; 300 children | Lesional/non-lesional AD skin plus matched controls | Transcriptomics linked to barrier, microbiome, and clinical data | Shows how microbiopsy can be embedded in longitudinal multiscale dermatology research. |
| Churiso 202012 | Microbiopsy; 29 cutaneous leishmaniasis suspects | Diverse Ethiopian cutaneous lesions | Real-time PCR and pain scoring | Showed lower pain than skin slit and confirmed all clinically suspected cases in a proof-of-principle diagnostic study. |
| Cloots 202113 | Microbiopsy; 201 participants across VL, PKDL, asymptomatic infection, and controls | Arm, neck, and PKDL lesion-edge skin | kDNA qPCR with parasite genome equivalents per microbiopsy | Linked skin parasite burden to disease status and transmission-oriented questions in human leishmaniasis. |
| Owen 202114 | Prospective VL-HIV cohort protocol; 91 planned patients | Serial day 0–29 skin, blood, and urine sampling | Blood/skin qPCR, buffy coat microscopy, urine antigen ELISA | Designs a microbiopsy-enabled workflow for diagnosis plus test-of-cure assessment in VL-HIV coinfection. |
| van Henten 202415 | Harpera microbiopsy subset within a larger 351-patient CL diagnostic study | Index lesion compared with skin slit, dental broach, and tape disc | PCR, diagnostic accuracy metrics, pain scores | Showed lower pain but workflow-dependent sensitivity, emphasizing processing and reference-standard effects. |
| Herrmann 202616 | Microbiopsy vs tape stripping vs skin scraping; 9 dogs | Axillary and inguinal skin in healthy and atopic dogs | RNA yield, RIN, RNA-seq quality control | Demonstrated excellent tolerability but poor microbiopsy RNA quality for transcriptomic use in veterinary skin research. |
| Yamada 202118 | Skin microbiopsy; fragile-skin pilot material | Outer forearm fragile skin and matched healthy-volunteer skin | Dermoscopy follow-up; targeted KRT5/KRT14 RT-qPCR normalized to GAPDH | Provides provisional perspective evidence suggesting acceptability and targeted gene-expression feasibility in fragile skin; not peer-reviewed and not used for performance claims. |
The analytical range reported across microbiopsy studies is unusually broad for such a small sampling device. DNA-oriented workflows have included direct extraction, Qubit quantification, whole-genome amplification, mutation screening by digital droplet PCR, and comparison with immunohistochemistry for molecular concordance.2,10 RNA-focused workflows have included RNAlater stabilization, PicoPure or column-based extraction, reverse transcription, standard-curve quantification, and relative gene-expression analysis by RT-qPCR.2,5,7 These assays have targeted structural skin markers (for example KRT14 and TYR), immune-cell markers (CD3E and CD19), and lesion-discriminating transcripts such as LINC00518 and GLI1.5,7
Imaging modalities have been equally important. Reflectance confocal microscopy and dermoscopy were used to characterize wound size, healing kinetics, and lesion topography in the engineering work.2 Confocal microscopy combined with live-cell probes such as CellROX and MitoSOX enabled functional assessment of oxidative stress in freshly collected microbiopsy tissue.8 Conventional histopathology remains central in comparative designs: shave or excisional biopsy still provides the morphological reference standard, while microbiopsy contributes high-specificity spatial molecular data that can be mapped against it.3,5,10 The platform therefore excels when paired methodologies are deliberate rather than mutually exclusive.
In infectious-disease applications, the dominant readout has been parasite-load qPCR rather than host-gene profiling alone. Leishmania studies quantified kinetoplast DNA from microbiopsy material and expressed results as parasite genome equivalents per microbiopsy, enabling direct comparison between skin and blood compartments and between arm, neck, and lesion-edge sampling sites12–14 ( Figure 5). This quantitative framing expands microbiopsy from simple positive/negative detection to a tool for burden estimation, spatial mapping, and treatment monitoring.
At the other end of the translational spectrum, Herrmann et al. used microbiopsy, skin scraping, and tape stripping to benchmark RNA concentration, RNA integrity, and RNA-seq quality control in companion-animal skin ( Figure 6).16 Their study is methodologically important because it shows that tolerability alone does not guarantee transcriptomic usability; low-yield microbiopsy RNA may still fail quality requirements even when sample collection itself is straightforward.
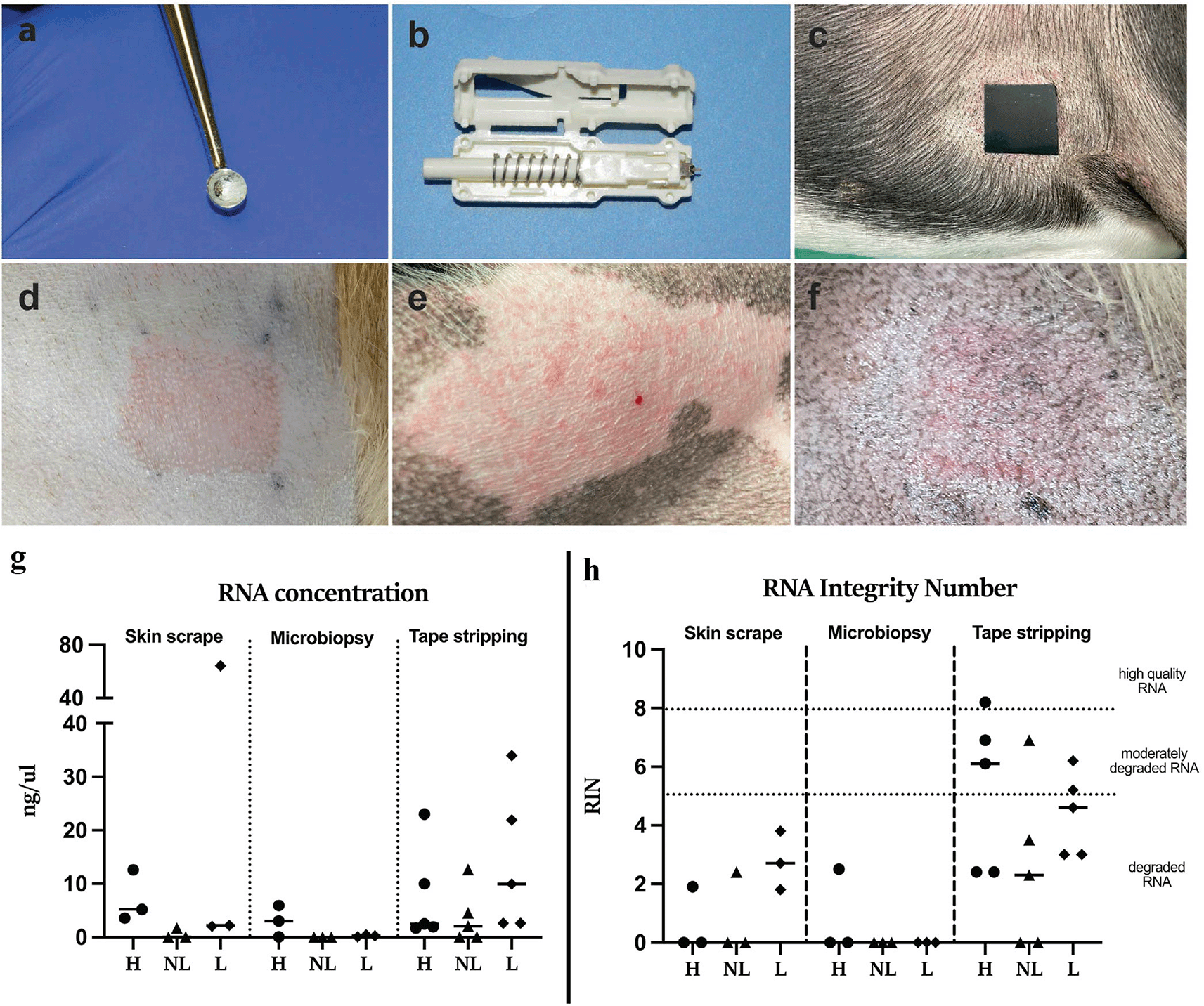
(Left) Comparative minimally invasive canine sampling methods and skin sites; (Right) RNA concentration and RNA integrity number across skin scraping, microbiopsy, and tape stripping. Source panels adapted from Herrmann et al.16
Most published microbiopsy studies have used statistical approaches appropriate to early translational or proof-of-concept work. Device-development studies relied on descriptive statistics, mean ± standard deviation summaries, one-way ANOVA with Tukey post-tests, and direct comparisons of DNA yield, pain score, or roughness parameters.2 RT-qPCR studies normalized expression using reference genes and the ΔCt framework, with replicate averaging and basic hypothesis testing.5,7 The sunscreen-toxicity study used paired designs and ANOVA to compare treatment conditions and control probes in a small volunteer cohort.8
The main statistical limitation is not analytical sophistication, but sample size and design heterogeneity. Many studies were intentionally small, lesion-specific, or method-development oriented.4,5,7,8,10 MAPS stands out because it explicitly plans adjusted regression analyses in a well-defined prospective cohort, linking microbiopsy transcriptomics to a much richer phenotypic context.9 For the field to mature, future microbiopsy research will need larger multisite cohorts, standardized endpoints, and reporting conventions that make between-study synthesis more reliable.
Larger diagnostic-accuracy studies have also introduced more explicit performance metrics. The 2024 Ethiopian comparison of dental broach, tape disc, and microbiopsy with PCR reported sensitivity, specificity, predictive values, receiver operating characteristic analyses, and alternative composite reference standards, demonstrating how strongly apparent device performance can depend on comparator choice and threshold setting.15
Table 2 provides an indicative comparison of commonly reported performance and feasibility measures. These values should not be interpreted as pooled estimates because device format, sampling site, stabilization, extraction chemistry, and downstream assay differed substantially across studies.
| Study/context | Device or comparator | Indicative yield or quality measure | Feasibility/interpretation | Evidence status |
|---|---|---|---|---|
| Lin 20132 | Original skin microbiopsy | Optimized geometry recovered DNA and RNA in the low-ng range; yield varied with channel width and application velocity. | Low pain scores and rapid visible recovery supported repeatable low-burden sampling. | Peer-reviewed proof-of-concept engineering study |
| Tom 20164 | Skin microbiopsy versus swab | HPV DNA could be detected from very small DNA inputs. | Smaller footprint improved positional specificity but sampled a narrower area than swabbing. | Peer-reviewed case report |
| Lei 20197 | Absorbent microbiopsy | Post-application dwell time increased RNA recovery and blood-associated signal. | Demonstrated simultaneous skin and blood capture, but workflow parameters strongly affected yield. | Peer-reviewed protocol study |
| Jain 202210 | Micro-punch biopsy devices | Whole-genome amplification supported BRAF V600E testing by ddPCR. | Genotyping agreed with reference testing in a small case series; larger validation is required. | Peer-reviewed case series |
| Herrmann 202616 | Canine microbiopsy, scraping, tape stripping | Microbiopsy and scraping yielded low-quality RNA for RNA-seq; tape stripping had higher RIN values but remained variable. | Excellent tolerability did not guarantee transcriptomic usability. | Peer-reviewed comparative feasibility study |
The most obvious limitation of microbiopsy is its size. Sample mass is deliberately small, which means analytical success depends on meticulous workflow control and sensitive downstream assays.2,7,10 Device performance is also protocol-dependent. In the original engineering study, collection efficiency changed markedly with channel width and application velocity, and DNA recovery was negligible at the lowest tested deployment speeds.2 In the absorbent format, simply holding the device in place for 10 seconds instead of releasing it immediately increased RNA recovery approximately fourfold.7 Thus, the technology is minimally invasive, but not methodologically forgiving.
A second limitation is interpretive context. Microbiopsy samples are molecularly rich, but they do not preserve tissue architecture and are therefore unsuitable for analyses requiring spatial localisation of immune infiltrates, vascular structures, or extracellular matrix organisation. This matters particularly in contexts such as dermal immune profiling, fibrosis, vasculitis, panniculitis, and other architecture-dependent inflammatory or fibrotic diseases. Sampling depth and molecular yield are variable, and RNA quantity is generally insufficient for unbiased transcriptomic discovery without pooling or extensive optimization, limiting many current workflows to targeted, hypothesis-driven analyses. Accordingly, microbiopsy should complement rather than substitute conventional biopsy in inflammatory, fibrotic, or architecture-dependent disease contexts. In pigmented lesions, for example, microbiopsy can provide discriminative gene-expression or mutation data, but histopathology remains necessary for definitive morphologic classification.5,10
Reference-standard selection is particularly important. In the large cutaneous leishmaniasis comparison, several patients who were negative by skin-slit PCR were repeatedly positive by one or more alternative sample types, leading the authors to recommend composite reference standards instead of single-test comparisons when benchmarking less invasive tools.15 This issue is highly relevant to microbiopsy literature because small spatial samples can appear discordant even when they are biologically informative.
Sample handling is a recurring practical bottleneck. Because input material is limited, pre-analytic losses matter disproportionately. Published studies have used dry ice, immediate stabilization, overnight lysis, RNAlater, modified extraction protocols, and whole-transcriptome or whole-genome amplification to protect data quality.2,7,9,10 These adaptations are effective, but they also increase workflow complexity and reduce direct comparability across studies. Standard operating procedures for dwell time, storage media, lesion annotation, extraction chemistry, batch effects, and quality-control thresholds will be essential if microbiopsy is to move from proof-of-principle use to multicentre deployment.
Recent veterinary transcriptomic work sharpens this point. Herrmann et al. found that canine microbiopsy and skin-scraping samples yielded low RNA quantities with poor integrity, whereas tape stripping produced the highest RNA concentration and RIN values (3.4–7.1) but still showed variability and suspected DNA contamination in downstream sequencing.16 The lesson is not that microbiopsy lacks translational potential, but that low-input RNA workflows remain extremely sensitive to stabilization, pooling strategy, storage conditions, extraction chemistry, and sample type. Routine single-device RNA-seq should therefore be regarded as unvalidated unless a given protocol provides explicit quality-control and reproducibility data.
The microbiopsy literature includes peer-reviewed engineering studies, case reports, diagnostic comparisons, protocols, and a small amount of author-provided or in-preparation material. To maintain critical distance, non-peer-reviewed material in this review is labelled as provisional or perspective evidence and is not used to support numerical performance claims. Where unpublished datasets are discussed, they should be retained only if their status is explicitly stated as exploratory, submitted, in preparation, or otherwise not yet peer reviewed.
The primary questions addressed by microbiopsy studies have evolved in a coherent sequence. Early work asked whether a sub-millimetre device could safely collect enough viable tissue for molecular analysis; the answer was yes, provided device geometry and deployment were optimized.2 Subsequent diagnostic studies asked whether the platform could detect clinically relevant targets with adequate spatial precision and analytical fidelity. HPV detection around warts, melanoma biomarker profiling, and BRAF genotyping all showed that microbiopsy can recover specific and clinically interpretable molecular information from lesions or matched control skin.4,5,10
Secondary outcomes have often been just as influential as the headline results. The engineering study generated practical insights about channel width, surface roughness, application velocity, pain, wound healing, and nucleic-acid quality that still inform later protocols.2 The wart study revealed that smaller sampling footprints can reduce false spatial spread compared with swabbing.4 The absorbent protocol demonstrated that dwell time is a recoverable performance parameter and that one device can be tuned toward either tissue-only or combined blood-and-skin outputs.7 The Jain case series showed that multiple samples can be banked from the same lesion for future use, adding a pre-analytic dimension that is highly relevant for translational biobanking.10 These secondary findings matter because they define the operational envelope of the technology. In a young field, knowing what changes yield, what improves specificity, and what can be repeated safely is often as important as the biological result itself.
A plausible future direction is the integration of microbiopsy-derived molecular data into multimodal skin research models. MAPS already illustrates part of this architecture: transcriptomics from microbiopsies linked to serial clinical examinations, skin-barrier measurements, microbiome sequencing, blood phenotyping, and environmental exposure data.9 In this setting, microbiopsy is best understood as one possible data-acquisition layer for longitudinal, hypothesis-driven skin biology rather than as a stand-alone route to digital-twin deployment.
The current evidence should be separated into three levels. Demonstrated uses include targeted DNA detection, RT-qPCR, mutation testing after amplification, and live-cell functional assays.2,4–8,10,12–15 Emerging uses include pooled or protocol-dependent transcriptomic workflows in carefully controlled cohort designs.9,16 Aspirational uses include routine single-device RNA-seq, clinical-grade multi-omics, and AI-enabled molecular twins. These latter applications are conceptually attractive, but they require validation of sampling reproducibility, analyte recovery, batch correction, computational interpretation, and clinical utility before they can be framed as established capabilities.
If transcriptomic protocols become more reliable, computational interpretation will become increasingly important. Cell-state deconvolution, pathway prioritisation, longitudinal trajectory analysis, and cross-cohort harmonisation could help convert serial low-input molecular snapshots into experimentally useful models of barrier dysfunction, inflammation, lesion evolution, or treatment response. For now, these analyses should be described as emerging or aspirational rather than inevitable.
Commercial and academic digital-biology platforms illustrate the type of informatics layer that might eventually sit downstream of validated microbiopsy datasets.17 However, such platforms should not be treated as evidence that microbiopsy-derived RNA-seq or multi-omic clinical interpretation is already routine. The immediate priority is still experimental validation: standardized collection, transparent quality metrics, comparator studies, and reproducible links between molecular readouts and biological or clinical outcomes.
Figure 7 illustrates a translational workflow for applying skin microbiopsy-based sampling, beginning with the clinical or research question and progressing through sampling design, laboratory processing, data interpretation, and integration into broader analytical frameworks. It highlights how microbiopsy approaches can support applications across diagnostic microbiology, oncology, functional modelling, and longitudinal studies, particularly where minimally invasive, site-specific, or repeated sampling is required.
Broader clinical adoption will depend on reducing workflow friction while improving assay validity. Published studies already show utility in virology, oncology, inflammation, parasitology, and exposure science.4–10,12–15 To move from selected pilot studies to routine deployment, future programs should standardize device nomenclature, establish validated sampling SOPs, define the circumstances in which microbiopsy adds value over swabs, tape strips, suction blistering, or direct biopsy, and report failures as well as successes. Automated extraction, multiplex nucleic-acid assays, and point-of-care-compatible processing could strengthen this translational pathway, but only where analytical and clinical validation are demonstrated.
Clinical expansion is most plausible in scenarios where repeatability, spatial precision, or cosmetic preservation matter: lesion triage before excision, molecular monitoring of topical treatment response, paediatric inflammatory dermatoses, and field studies in resource-limited settings.6,7,9,10,12–15 The platform may never replace punch biopsy across dermatology, but it does not need to. Its clinical role is likely to be niche-defining rather than universal.
At the same time, expansion should be evidence-led rather than assumption-led. Comparative diagnostic studies in cutaneous leishmaniasis and comparative-medicine data from dogs show that microbiopsy will not automatically be the best low-burden method for every molecular endpoint; in some settings it may outperform conventional sampling on pain and spatial precision, while in others tape stripping or alternative minimally invasive tools may yield better RNA or diagnostic performance.11,15,16 The most likely future is therefore a complementary one, in which microbiopsy is deployed where viable sub-surface material and repeatability are decisive.
A further opportunity lies in microbiopsy biobanking. Because the device can collect serial lesional, perilesional, and control samples with relatively low burden, it is well suited to repositories built around temporal and spatial molecular context rather than bulk tissue volume.7,9,10 Banked microbiopsies could be paired with digital images, dermoscopy, barrier metrics, conventional pathology, treatment timelines, perturbation exposures, and longitudinal outcomes. Such repositories would be valuable for biomarker discovery, assay validation, and retrospective cohort re-analysis, provided the limitations of low-input material are captured in the metadata.
To realize this potential, harmonized metadata will be essential. Storage conditions, stabilization media, dwell time, lesion site, device format, operator training, and downstream assay compatibility should all be captured at the point of sampling. Biobanks built on tiny specimens succeed or fail on annotation quality. Microbiopsy makes such banks realistic; data governance, quality standards, and fit-for-purpose endpoint selection will determine whether they become useful.
Clinical translation will require attention to regulatory classification, intended use, analytical validation, and operator training. A device used for research sampling, investigational biomarker development, or clinical diagnosis may require different levels of evidence and oversight. Diagnostic claims should therefore be benchmarked against appropriate clinical reference standards, with transparent reporting of sensitivity, specificity, indeterminate results, adverse events, sampling failures, and patient-reported burden.
Ethical reporting is also important because several authors in the field have contributed to microbiopsy device development. Manuscripts should disclose intellectual-property, commercial, consultancy, funding, and device-development relationships; clearly label adapted figures and confirm permissions; and distinguish peer-reviewed evidence from author-provided or exploratory material. These steps strengthen rather than weaken the review by making the technology’s current evidence base explicit.
Skin microbiopsy has evolved from a proof-of-concept sub-millimetre device into a minimally invasive sampling platform that can support targeted DNA analysis, RT-qPCR, live-cell functional assays, infectious-disease parasitology, and longitudinal cohort designs.2,4–10,12–16 Its value lies not in replacing established methods, but in occupying an intermediate position between surface sampling and conventional biopsy. Where swabs and tape strips may lack viable sub-surface material, and where punch biopsy imposes unacceptable burden for repeated or spatially targeted use, microbiopsy can provide a low-morbidity route to selected molecular and functional readouts.1,2,7,10,11
Challenges remain substantial. The evidence base is still weighted toward method-development studies, case reports, protocols, and small translational applications. Sample mass is inherently limited; tissue architecture is not preserved; RNA yield and integrity vary; and endpoint-dependent variability must be addressed through rigorous standardization. Robust benchmarking against tape stripping, swabbing, suction blistering, scraping, and conventional biopsy will be essential, particularly for inflammatory, fibrotic, infectious, and architecture-dependent disease contexts. Non-peer-reviewed author-provided material should remain clearly labelled as provisional until publication status is confirmed.
This review has aimed to provide both a clear account of how the field has developed and practical guidance on when microbiopsy is fit for purpose. The most defensible conclusion is complementary rather than substitutive: microbiopsy should be chosen when the research question specifically benefits from spatial precision, repeatability, minimal visible morbidity, or access to viable sub-surface material. Future work should define standardized workflows, comparator-specific performance, regulatory pathways, and transparent reporting standards before broader clinical or industrial use is assumed.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.

Comments on this article Comments (0)