Keywords
Helicobacter pylori, cagA, MGMT promoter methylation, bisulphate, epigenetics, qPCR.
This article is included in the Fallujah Multidisciplinary Science and Innovation gateway.
Helicobacter pylori, cagA, MGMT promoter methylation, bisulphate, epigenetics, qPCR.
Gastric cancer continues to be a significant contributor to cancer morbidity and mortality, with Helicobacter pylori identified as a primary upstream factor in chronic gastritis and gastric carcinogenesis. In addition to inflammation, growing evidence suggests that epigenetic remodeling, especially DNA methylation of promoter regions, serves as a mechanistic link between infection and malignant transformation.1,2 DNA methylation at CpG islands can turn off genes that stop tumors from growing and make it harder for the genome to stay stable. O-6-methylguanine-DNA methyltransferase (MGMT) is an important DNA repair enzyme that removes O-6 alkyl adducts.3,4 When MGMT is hypermethylated at its promoter, it makes it harder for cells to repair DNA and increases the number of mutations. Virulence factors of H. pylori, particularly cagA, are biologically equipped to enhance host signaling and oxidative stress, promoting abnormal methylation in the gastric epithelium.5,6 In high-burden contexts, elucidating the correlation between H. pylori (and CagA +/-) and MGMT promoter methylation across the clinical continuum from gastritis and peptic ulcer disease to lymphoma and adenocarcinoma is of immediate translational significance.7,8
This study aims to elucidate the relationship between H. pylori infection, specifically CagA-positive strains, and MGMT promoter hypermethylation, as well as to assess the variability of methylation frequencies across standardized gastroduodenal diagnoses. The focus is on quantifying effect sizes for infection–methylation associations and contextualising methylation within the observed disease gradient, while also analyzing prevalent cofactors such as smoking, sex, residence, chronic illness, and treatment.9
The primary objective is to assess the correlation between H. pylori cagA positive and MGMT promoter hypermethylation within the gastroduodenal disease spectrum in an Iraqi cohort. The goals are: (i) to find out how common H. pylori, cagA carriage, and MGMT hypermethylation are in both control and patient groups; (ii) to use the right inferential statistics to find out how strong the link is between infection status and MGMT hypermethylation; (iii) to describe how MGMT methylation is spread across acute and chronic gastritis, peptic ulcer disease, gastric lymphoma, and gastric adenocarcinoma; (iv) to look into links between methylation status and key cofactors, pointing out variables that show strong links; and (v) to evaluate the potential of MGMT promoter hypermethylation as an epigenetic biomarker to help with risk stratification and early detection in endoscopy pathways.
The study included 120 participants undergoing oesophago-gastroduodenoscopy (OGD): 40 endoscopy-verified controls without gastroduodenal disease and 80 patients diagnosed with gastritis, peptic ulcer disease, gastric lymphoma, or gastric adenocarcinoma. For each participant one biopsy was used immediately for the rapid urease test. The remaining biopsies were placed into sterile, labelled microtubes and transported on ice. The distributions of age and sex were broadly similar between cases and controls.
The inclusion criteria were individuals aged 18 years or older, possessing sufficient gastric biopsy material, and having comprehensive clinical data; the control group exhibited a normal OGD without any macroscopic or histological evidence of gastroduodenal disease.
Exclusion criteria included those with history autoimmune diseases and other types of cancers.
Endoscopic biopsies were collected following standard OGD protocols and promptly processed for Helicobacter pylori testing and nucleic acid workflows. The rapid urease testing was used at the bedside using medium from the manufacturer (Himedia, India). For culture, biopsies were transported in Stuart medium and plated on blood agar supplemented with Skirrow selective supplement and 5% defibrinated sheep blood (Himedia, India); plates were incubated micro aerobically at 37°C for 3 days. Colonial morphology, Gram stain, oxidase, catalase, and urease tests were used to diagnose presumptive H. pylori colonies. Kligler iron agar was used when necessary to rule out Enterobacteriaceae. Biopsy samples DNA extraction was stored at −20°C. The silica-column based DNA extraction kit (G-spin DNA extraction kit, iNtRON Biotechnology, Korea) was used to isolate bacterial DNA from a biopsy or culture. Using primers made by Macrogen (Korea), CagA-F 5′-GATAGGGATAACAGGCAAGC-3′ and CagA-R 5′-GGGGGTTGTATGATATTTTC-3′ (amplicon size 297 bp),10 the study used conventional PCR to assess the virulence of H. pylori. The reactions were prepared with Maxime PCR PreMix (i-Taq; iNtRON, Korea) in 25 μL and cycled on a SimpliAmp™ thermal cycler (Applied Biosystems, USA) with 94°C 5 min; 35 cycles of 94°C 35 s, 50°C 35 s, 72°C 35 s; final extension 72°C 7 min. RedSafe to stain 2% agarose/TBE gels were used to visualize the PCR products. A 100-bp ladder was used as a size marker.
The study used the Monarch® Genomic DNA Purification Kit (New England Biolabs, USA) to clean up genomic DNA from gastric tissue. The DNA concentration and purity were assessed using Nanodrop, Thermofisher scientific, USA, by measuring absorbance of samples with an OD 260/280 ratio between 1.8 and 2.1 and concentration ranged from 122 to 400 pg/ul were considered suitable for downstream bisulfite conversion and MSP. The MethylEdge® Bisulfite Conversion System (Promega, USA; Cat. N1301) was used to convert bisulfite, following the manufacturer’s instructions, which included desulphonation and spin-column cleanup. We used methylation-specific PCR (MSP) to check the status of the MGMT promoter. The primers were used Macrogen (Korea) for the methylated set MGMT-M (Left-M 5′-TATTTTTGTGATAGGAAAAGGTACG-3′; Right-M 5′-TAAAACAATCTACGCATCCTCG-3′; 191 bp) and the unmethylated set MGMT-U (Left-U 5′-ATTTTTGTGATAGGAAAAGGTATGG-3′; Right-U 5′-CTAAAACAATCTACACATCCTCACT-3′; 191 bp). MSPs were performed on the SimpliAmp™ platform under optimized cycling conditions, incorporating fully methylated and unmethylated controls in each batch. Real-time confirmation utilized SYBR Green PCR Master Mix (Synthol, Russia) in 20 μL reactions (10 μL master mix, 1 μL MgCl2, 1 μL each primer, 2 μL template, 5 μL nuclease-free water) and the specified program: 95°C for 5 minutes, followed by 45 cycles of 95°C for 20 seconds, 53–60°C for 20 seconds, and 72°C for 20 seconds. Melt-curve analysis confirmed specificity.
The study comprised 120 participants (40 controls and 80 patients). The sex distribution did not exhibit a significant difference between groups (females: 32.5% in controls vs 43.8% in patients; χ2 = 1.406, p = 0.236; OR = 0.62, 95% CI 0.28–1.37). Patients were aged greater than controls (42.83 ± 13.49 vs 39.95 ± 12.59 years), but there are no significant differences between the two groups (χ2 = 50.411, p = 0.125; OR = 2.33, 95% CI 1.04–5.22). There was a significant difference in residence by group: 50.0% of patients lived in rural areas compared to 30.0% of controls (χ2 = 4.344, p = 0.037). The complementary comparison showed that patients were less likely to live in urban areas (χ2 = 6.018, p = 0.014; OR = 0.38, 95% CI 0.17–0.83). Patients had a lower rate of smoking than controls (41.2% vs. 65.0%), but the difference was statistically significant (χ2 = 13.125, p = 0.001). Patients were more likely to receive therapy (treated: 46.3% vs 27.5%; χ2 = 3.906, p = 0.048; OR = 2.27, 95% CI 1.00–5.16). By design and clinical adjudication, all controls were free of gastroduodenal disease (100.0%), while all patients had a documented diagnosis (χ2 = 120.0, p < 0.001) as reported in Table 1.
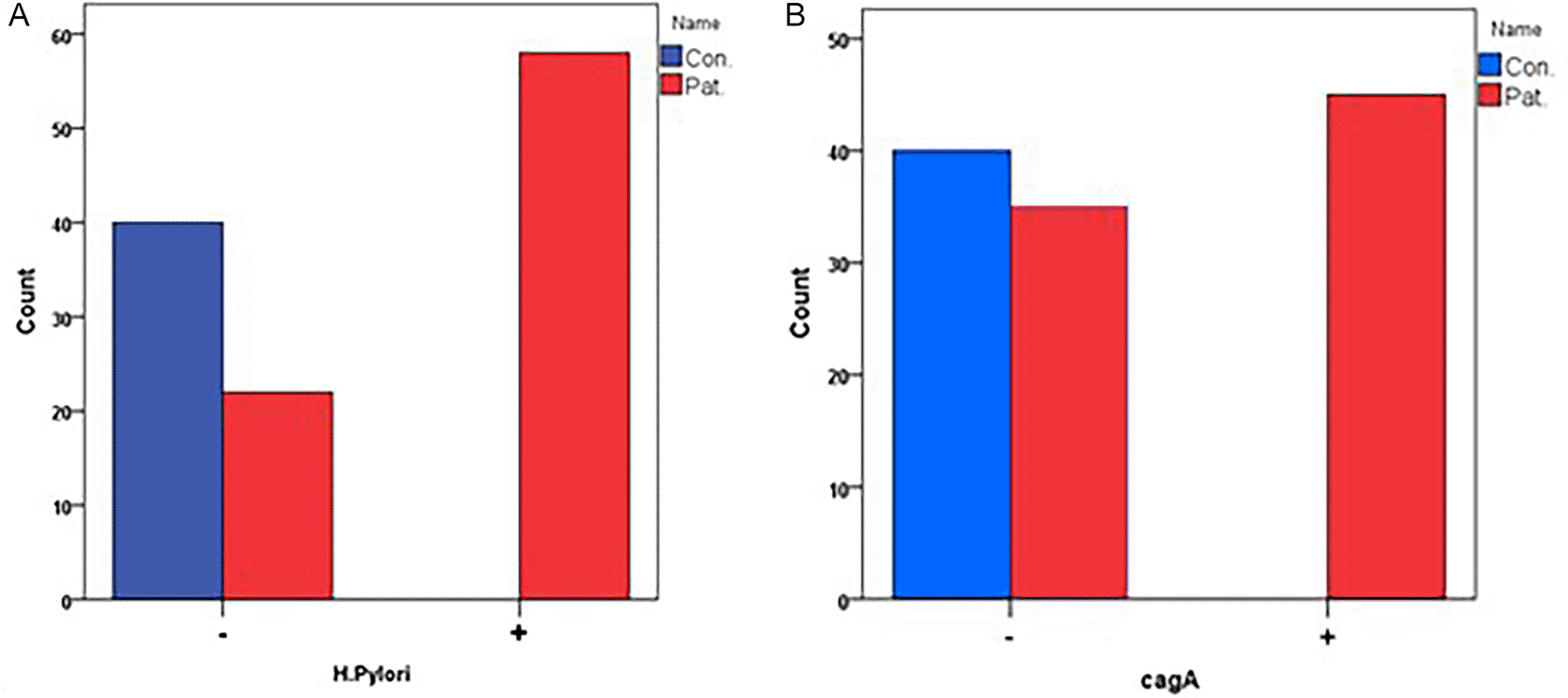
There was a clear difference in the number of infections and the number of virulence carriers between the groups. The prevalence of Helicobacter pylori was significantly higher in patients (72.5%) and there is no infection recorded in control (0%) with χ2 = 56.129 and p < 0.00, indicating substantially elevated odds of infection in patients (OR = 210.60, 95% CI 12.42–3572.22). The cagA virulence gene was also found in more than half of the patients but not in any of the controls (56.3% vs 0%; χ2 = 36.000, p < 0.001), which means That the effect size was very large (OR = 103.82, 95% CI 6.17–1747.38) as demonstrated in Figure 1 A+B.

There was a clear difference between the two groups: hypermethylation was present in 66.3% of patients and 0% of controls (χ2 = 47.463, p < 0.001), which is a large effect size (OR = 157.58, 95% CI 9.33–2661.18). As reported in Table 2.
In a sample of 120 participants, sex, residence, chronic disease, and therapy exhibited no significant correlations with MGMT promoter methylation in bivariate analysis (all p > 0.10). The distribution of females versus males was similar between hypermethylated and unmethylated cases (χ2 = 0.681, p = 0.409). The status of chronic disease did not vary with methylation (χ2 = 0.510, p = 0.475). Residence indicated a non-significant trend towards increased hypermethylation in rural compared to urban environments (χ2 = 2.239, p = 0.135). Previous treatment did not correlate with methylation status (χ2 = 0.456, p = 0.499). Conversely, smoking exhibited a significant correlation with methylation (overall χ2 with 3 categories = 15.218, p < 0.001), and the aggregated comparison revealed increased odds of hypermethylation among smokers compared to non-smokers (OR = 4.55, 95% CI 1.96–10.55) as showed in Table 3.
There was a significant association between H. pylori infection and MGMT promoter hypermethylation (Table 4). In H. pylori positive cases, 74.1% exhibited hypermethylation, whereas only 16.1% of H. pylori–negative cases showed this characteristic (χ2 = 40.892, p < 0.001), indicating significantly elevated odds of hypermethylation associated with infection (OR = 14.91, 95% CI 6.09–36.60). The association was more significant for cagA positivity, with hypermethylation seen in 91.1% of cagA-positive people compared to 16.0% of cagA-negative people (χ2 = 64.345, p < 0.001; OR = 53.81, 95% CI 16.24–178.31) as reported in Table 4.
Helicobacter pylori infection significantly promotes the progression of chronic gastritis to gastric carcinoma by influencing inflammatory and oxidative pathways, as well as inducing epigenetic modification of host gastric epithelial cells. The present study has a cross-sectional, observational design and therefore cannot establish a definitive causal or mechanistic relationship between H. pylori, particularly cagA-positive strains, and MGMT promoter hypermethylation. The findings demonstrate H. pylori induced oxidative stress, DNMT upregulation, and promoter hypermethylation. Integrating insights that H. pylori act as an upstream driver of MGMT epigenetic silencing, but longitudinal and interventional studies are required to confirm this causal pathway. When tumor suppressors and DNA repair genes, like MGMT, get too much methylation, the DNA repair machinery stops working. This is the most important change.11 This makes mutations build up and the cells become cancerous. recurrent inflammation, oxidative stress, and host signaling pathways are the main reasons why H. pylori cause MGMT to be epigenetically silenced. Studies show that when H. pylori invade the gastric mucosa, it triggers a strong immune response that damages DNA and turns on DNA damage response pathways.12 This leads to an overexpression of DNA methyltransferases (DNMTs). These enzymes are very important because they move methyl groups to cytosine residues. This leads to unusual DNA methylation in the promoter areas of genes that control cell cycle arrest, apoptosis, and DNA repair. MGMT, a crucial DNA repair gene, is particularly susceptible to methylation-mediated silencing during H. pylori infection, promoting neoplastic progression.13 The detection of MGMT promoter hypermethylation in inflamed but histologically benign mucosa underscores its possible function as an early molecular event in gastric carcinogenesis. Longitudinal data suggest that the elimination of H. pylori infection can partially reverse or impede these methylation changes, indicating that bacterial eradication not only reduces inflammation but may also restore normal epigenetic regulation in the gastric mucosa.14 A number of studies suggest that MGMT promoter hypermethylation may serve as a non-invasive biomarker for risk assessment and early detection, potentially influencing therapeutic sensitivity and prognosis in cancers associated with MGMT silencing.15 Nonetheless, the strength of this relationship may vary among populations due to genetic, dietary, and environmental influences on methyl group metabolism and inflammatory response, supporting the conclusions of this study.16,17 A study confirmed the results of this research, demonstrating that H. pylori colonisation correlates with elevated DNMT activity and a hypermethylation of islands of CpG in the gastric mucosa, with MGMT identified as one of the most frequently silenced DNA repair genes.18 A study confirmed these findings by showing that MGMT methylation can be detected not only in cancerous tissues but also in inflamed, non-neoplastic mucosa, corresponding with an epigenetic “field defect” that occurs prior to histological transformation. Another study confirmed the finding of this study that cagA positivity is linked to the strongest association. This study found that cagA-positive strains cause more significant promoter hypermethylation and greater suppression of MGMT expression than cagA-negative strains.19 A subsequent study validated the present findings by demonstrating that H. pylori-induced oxidative and inflammatory signaling elevates DNMT1 expression and encourages MGMT silencing, thereby hindering the repair of O6-alkylguanine adducts and promoting the accumulation of mutations. The gradient hypermethylation identified in gastritis and peptic ulcer disease, progressing to malignant outcomes, is supported by prior clinical studies.20 A study validated the results of this research, illustrating progressive elevations in MGMT methylation from chronic gastritis to intestinal metaplasia and carcinoma.21,22 This suggests that methylation acts as an early and enduring indicator of H. pylori-associated mucosal remodeling. A study confirmed a similar trend by showing that MGMT methylation often comes before morphological dysplasia, which supports its use as a risk-stratification marker during endoscopic surveillance.23 Interventional data supports our etiological interpretation: a study confirmed the findings of this research and recorded a partial reversal or reduction of aberrant methylation following H. pylori eradication, suggesting that a segment of the MGMT methylation is infection-dependent and subject to modification. A study validated the results of this research demonstrating a heightened probability of MGMT hypermethylation in smokers, consistent with cumulative oxidative stress and methylation pressure.24 A study also confirmed that dietary methyl donors, nitrosamine exposure, and polymorphisms in methylation or repair genes influence the variations in effect sizes across studies.25 In addition to MGMT, a study confirmed the broader context and documented the coordinated methylation of additional tumor-suppressor and mismatch-repair loci in H. pylori–infected mucosa, situating MGMT within a more extensive, infection-induced epigenetic framework that undermines genome preservation and epithelial homeostasis.26 The comparative evidence collectively supports a unified model in which H. pylori, especially the most cagA positive strains, act as an upstream catalyst for MGMT promoter hypermethylation during the initial phases of the gastritis carcinoma sequence. Studies validating these results indicate a persistent increase in MGMT methylation within infected, inflamed mucosa, its presence in precancerous phases, and a degree of reversibility following eradication.26,27 In contrast, research that challenges these findings primarily focusses on late-stage, tumor-exclusive cohorts, where subsequent epigenetic remodeling may obscure the initial microbial signal. In this context, MGMT hypermethylation is recognized as an indicator of infection-related epigenetic damage and a potential biomarker for risk stratification in endoscopic procedures.27 Standardized, quantitative methylation assays performed longitudinally across well-defined stages accounting for cagA status, sampling site, eradication history, and lifestyle cofactors are imperative to address existing discrepancies and to determine the clinical relevance of MGMT methylation in H. pylori-associated gastric carcinogenesis.28 The extremely large ORs and wide CI for some comparisons (e.g., infection prevalence in cases vs controls) result from zero events in the control group. These estimates indicating the presence of a very strong association rather than providing an exact quantitative measure of risk.
In conclusion, the interaction of inflammatory, oxidative, and virulence-mediated mechanisms clarifies the strong link between H. pylori infection, especially with cagA-positive strains, and MGMT promoter hypermethylation in the gastric mucosa. This epigenetic alteration serves as a crucial connection between chronic infection and the molecular initiation of gastric carcinogenesis by disrupting an essential DNA repair mechanism. MGMT hypermethylation not only indicates the degree of damage inflicted by infection but also functions as a prognostic marker for malignant potential. Its frequent occurrence across diverse populations and disease stages underscores its generality as an epigenetic signature linked to infection. Comprehending and targeting this methylation pathway offers promising opportunities for early diagnosis, chemoprevention, and customised management of H. pylori-related gastric cancer.
This study was conducted in accordance with the ethical standards of the Iraqi Ministry of Health and the regulations of the Al-Anbar Health Directorate. Ethical approval was obtained from the Research Committee of the Al-Anbar Health Directorate (Approval No. 35813, dated 28 October 2025). In addition, ethical approval was granted by the Research Ethics Committee of the University of Fallujah, College of Applied Sciences (Approval No. AS-EC/0011, dated 21 December 2025). All procedures involving human participants were performed in accordance with the Declaration of Helsinki. Gastric biopsy samples were collected only after obtaining written informed consent from all participants. All personal identifiers were removed prior to data analysis to ensure participant confidentiality.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)