Keywords
Thalassemia, genetic variations, in silico, amino acid, molecular technique
This article is included in the Genomics and Genetics gateway.
Thalassemia, genetic variations, in silico, amino acid, molecular technique
Thalassemia is one of the most common monogenic hemoglobinopathy disorders worldwide as a result of inbreeding that started in the Mediterranean region. This disease was first recognized in 1925 by an American doctor named dr. Thomas Benton Cooley, whose name is used for this disease, which is Cooley's Anemia.1 Currently there are more than 350 genetic variations of thalassemia are observed and reported. This disease is inherited in an autosomal recessive manner following Mendelian rules. Clinically, thalassemia is divided into thalassemia major, thalassemia intermedia, and thalassemia minor.2
Individuals with thalassemia have various clinical symptoms, including pale skin color, fatigue and weakness, dark urine production, irregular heartbeat, irregular menstrual schedule, hepatosplenomegaly, pale conjunctiva, malocclusion of the maxillary bone, and stunted growth.3,4 Thalassemia can be diagnosed early by performing a Complete Blood Count (CBC) or hemoglobin test. The Complete Blood Count test measures the amount of hemoglobin and various types of blood cells, such as red blood cells, from an individual. Individuals with thalassemia have a lower red blood cell (RBC) volume than normal individuals.5 Determining genetic variations in thalassemia subjects also plays an important role, but has not been extensively studied in Indonesia.
Worldwide, it is estimated that approximately 320.000 babies are born with hemoglobinopathies each year, and 80% of these occur in low- or middle-income countries.4 Indonesia, one of the countries located along the “Thalassemia Belt, is home to more than 260 million people. Approximately 5% of this population is thought to carry the thalassemia gene, also known as the β-globin gene. With a birth rate of approximately 20% annually, according to Wahidiyat,3 there are approximately 2.500 new thalassaemia major cases per year, making Indonesia a hot spot for thalassemia. This study aimed to provide mutation analysis data for thalassemia subjects in Indonesia. Based on the data obtained from this study, we also provide a bioinformatics analysis of amino acid interactions from the variants found.
Twenty participants were recruited for this study from the Thalassemia Center on Cipto Mangunkusumo Hospital (RSCM), Central Jakarta, DKI Jakarta, Indonesia. All the participants already signed an assent form. The content of informed assent is being explained verbally to the participant. Informed assent was written in paper and signed by the participant under supervision from the parents and the researcher team. This study was approved by the Ethics Committee of the Faculty of Medicine, Universitas Indonesia (No. KET-600/UN2.F1/ETIK/PPM.00.02/2023). Blood samples were collected in ethylenediaminetetraacetic acid (EDTA) tubes. Blood samples were collected from thalassemia subjects by collecting 3 mL of blood before transfusion. The minimum time interval between the last transfusion and sample collection was 2 weeks. The samples were stored at 4 oC until genomic DNA (gDNA) extraction.
Raw samples (515 μL) were placed in a 1.5 ml microtube. A volume of 930 μL of RBC Lysis Buffer reagent (Geneaid Biotech Ltd., Taiwan)6 was added to the sample. The samples were then incubated for 19 minutes. The microtubes were then centrifuged (Eppendorf 5424R, Germany) at 3,000 g for 5 min. A white precipitate formed at the bottom of the microtubes. The supernatant was then discarded. A volume of 100 μL of RBC Lysis Buffer (Geneaid Biotech Ltd, Taiwan) and 200 μL of GB Buffer were added (Geneaid Biotech Ltd, Taiwan) and mixed thoroughly by shaking the microtubes. The samples were incubated for 11 min at 65 oC heater block (Stuart SBH130D, England) and then transferred to a new sterile GD Column. Samples were then centrifuged in an Eppendorf 5424R (Germany) for 5 min at 15,000 × g (room temperature), and the supernatant was transferred to a fresh, sterilized microcentrifuge tube. A volume of 400 μL W1 Buffer (Geneaid Biotech Ltd., Taiwan) was added. The samples were then centrifuged in an Eppendorf 5415R centrifuge at 15,000 × g (room temperature) for 1 min at room temperature. A volume of 600 μL of Wash Buffer (Geneaid Biotech Ltd, Taiwan) was added. The samples were then centrifuged (Eppendorf 5415R) at 15,000 × g (room temperature), again for 1 min. After replacing the GD column with a fresh collection tube, 100 μL of Elution Buffer (Geneaid Biotech Ltd, Taiwan) was added carefully, and the sample was incubated for 3 min. After 3 min of incubation, the tubes were centrifuged (Eppendorf 5415R) for 1 min at 15,000 × g (room temperature). Genomic DNA (gDNA) samples were frozen at −20°C for further use.
Genomic DNA concentration and purity were quantitatively assessed using a spectrophotometric assay with a Varioskan microplate reader (Thermo Fisher Scientific, United States).7 Absorbance was measured at wavelengths of 260 and 280 nm (A260 and A280, respectively). The absorbance quotient (OD260/OD280) was used to estimate the DNA purity. An absorbance quotient ratio between 1.8 and 2.0 was considered good for the purified gDNA. A ratio <1.8 indicated protein contamination, while a ratio of >2.0 indicated RNA contamination.
The gene used in this study was β-globin (NM_000518.5), located on chromosome 11p15.5, which consists of three exons. The FASTA sequence of the gene was obtained from NCBI (https://www.ncbi.nlm.nih.gov/gene/3043), whereas the FASTA coding sequence (CCDS) was obtained from Ensembl! (https://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi?REQUEST=CCDS&DATA=CCDS7753). All FASTA sequences were annotated using Benchling (https://www.benchling.com/), primers for polymerase chain reaction (PCR) were designed, and the quality was assessed using NetPrimer (http://www.premierbiosoft.com/NetPrimer/AnalyzePrimer.jsp) and NCBI Primer Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/).
PCR for the β-globin gene was performed using the following primer pair: forward 5’AGGTACGGCTGTCATCACT’3 and reverse 5’ TGGACTTAGGGAACAAAGG’3. A PCR mix of i-MAX II (iNTRON Biotechnology) was used. The PCR program consisted of pre-denaturation at 94°C for 2 min, denaturation at 94°C for 30 s, annealing at 58°C for 30 s, extension at 72°C for 55 s, repeated for 35 cycles, and a final extension at 72°C for 10 min. Agarose gel electrophoresis was performed to verify the quality of PCR products. The PCR products were separated on a 0.8% agarose gel at 100 volts for 45 min (Bio-Rad, United States). The gels were visualized using a gel documentation system (Accuris Instruments).8
Sequencing result obtained was analyzed using FinchTV version 1.4 (downloaded from https://finchtv.software.informer.com/1.4/#google_vignette) and BioEdit version 7.2 (downloaded from https://bioedit.software.informer.com/7.2/) software to align its genes. Wild-type FASTA sequences were obtained from the NCBI database (https://www.ncbi.nlm.nih.gov/nuccore/NG_059281.1?report=fasta&from=5001&to=6608). Computational and predictive data were obtained by assessing the clinical significance of each variant using Varsome consensus scoring (https://varsome.com/). The classification is divided into pathogenic, likely pathogenic, uncertain significance (VUS), likely benign, and benign. The effects of each variant on amino acid interactions, bonding, and side chains were analyzed using Biovia Discovery Studio 24 (downloaded from https://discover.3ds.com/discovery-studio-visualizer-download ). The wild-type amino acid sequences were obtained from the Alphafold Protein Structure Database (https://alphafold.ebi.ac.uk/search/text/HBB). The wild-type sequence of the β-globin gene was imported to Biovia Discovery Studio 24 and the mutation point on the amino acid sequence was annotated to analyze the change in its interactions.
This study reported all results from 20 respondents who had been diagnosed with β/β thalassemia and β/HbE thalassemia. Respondents recruited for this study were aged <2 to >10 years. The majority of the tribes from all respondents were Javanese, Sundanese, and Betawinese. One respondent was Chinese. According to Table 1, 10 genetic variations were observed in the Sanger Sequencing analysis. Several variants are classified as intronic variants that disrupt the splice site, whereas the others are missense, nonsense, and frameshift variants. Mutation found in this study is reported using its common name alongside with its nomenclature according to Human Genome Variation Society (HGVS). The most common variants found in this study is the Cd26 (HGVS: c.79G>A; p.(Glu27Lys)) and IVS1nt.5 (HGVS: c.92+5G>C; p.(?)) This is partly in accordance with a study by Wahidiyat et al. (2022). This implies that both Indonesian and Melanesian people are likely to share a common origin.
All variants observed in this study were classified as pathogenic, as confirmed by Varsomes and ClinVar. Missense variants reported in this study consist of Cd26 (c.79G>A) and Cd30 (c.92G>A), which are known to have an impact on amino acid translation from mRNA, leading to a change from glutamine at position 27 to Lysine and Arginine at position 31 to threonine. The nonsense variant reported in this study is c.47G>A, which leads to a premature stop codon starting at amino acid position 16. Nonsense variants are predicted to cause greater severity in thalassemia subjects than in those with missense variants only.
The most common variant, IVS1nt.5 (c.92+5G>C), is an intronic pathogenic variant of β-globin gene. Functional analysis using Human Splicing Finder showed that this variant loses the canonical splicing donor site, which consequently leads to loss of the start codon (ATG) and excision of exon 1 from the open reading frame (ORF). Owing to the altered mRNA splicing pattern, this variant encodes a shortened mRNA and may result in the loss of protein expression. Other intronic variants observed in this study are also predicted to disrupt the splicing site, leading to clinical observations in thalassemia subjects.9
The genotypes of the thalassemia subjects recruited for this study are summarized in Table 2. The most frequent genotype observed was HbE/IVS1nt.5 (HGVS: c.79G>A/c.92+5G>C). followed by homozygous variant of IVS1nt.5/IVS1nt.5 (HGVS: c.92+5G>C/c.92+5G>C). Referring to a previous study by Wahidiyat et al. (2022),10 all variants identified including IVS1nt.5 (HGVS: c.92+5G>C), IVSIInt.654 (HGVS: c.316-197C>T), Cd26 (HGVS: c.79G>A); IVS1nt.1 (c.92+1G>T). According to Sanger sequencing analysis, this study also discovered rare mutations in Indonesian populations, such as Cd35 (HGVS: c.110delC; p.(Pro37Leu)), and reported ClinVar as a pathogenic variant.
The initial study on 23 thalassemia subjects in Northern Vietnam using sequence analysis also mainly detected c.79G>A, c.124-127delTTCT, and c.52A>T, in addition to an indigenous mutation in Vietnam, c.287_288insA.11 Recently, a small report from 22 β-thalassemia subjects in Central Vietnam showed that the three mutations c.79G>A, c.52A >T, and c.124_127delTTCT accounted for 29.2%, 25.0%, and 18.8% of the mutant alleles, respectively.12 However, it should be noted that in Myanmar,13 Thailand,14 and China,15 the most common mutation was c.124-127delTTCT, not c.79G>A, similar to the Indonesian population.
The genotype of Indonesian thalassemia subjects mostly consisted of compound heterozygous variants, including missense/missense, intronic/missense, missense/nonsense, intronic/frameshift, and missense/frameshift. Only a small number of subjects are known to have homozygous variants. However, all subjects received a 2 weeks blood transfusion with an Hb level < 9 g/dL. In this study, we concluded that every variant causes a severe type of thalassemia.
The term of “rare” in a genetic variant is defined as a variant that is not commonly discovered in a specific population. According to the results of Sanger Sequencing, there are nucleotide changes that lead to frameshifts in nucleotide reads, as shown in Figure 1. This novel variant showed changes in the coding sequence (CDS) position of 110. Changes in nucleotides are deletions of cytosine and frameshift of thymine, located immediately after the nucleotide at position 110 (HGVS: c.110delC; p.(Pro37Leufs*23)). This also caused frameshift nucleotide reads in the rest of the sequencing results. These changes manifested in amino acid changes from proline at position 37 to leucine, and a premature stop codon occurred after 23 amino acid translations. The classification of all variants reported, including this frameshift variant, was based on the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology.16
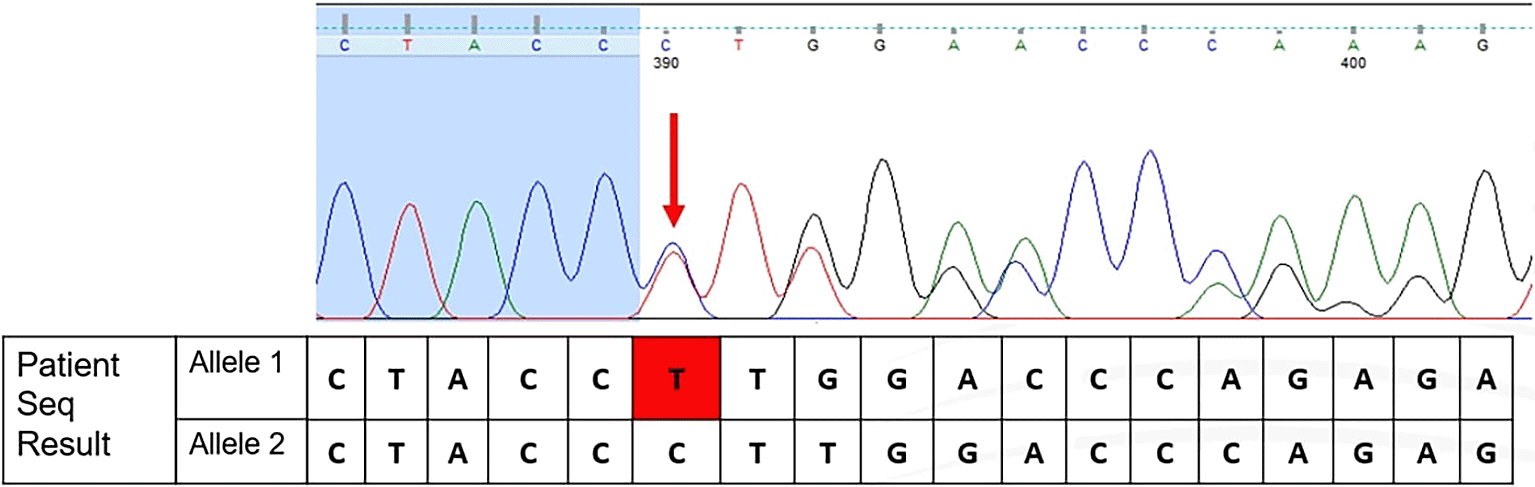
According to Rujito et al. (2015),17 only two of 209 subjects were observed to have this mutation. This frameshift variant was also found in Malay populations. The genotype observed by Rujito et al. (2015)17 was compound heterozygous with IVS1nt.5 (HGVS: c.92+5G>C).17 However, this study found that this variant was observed in Cd26 (HGVS: c.79G>A). The frameshift variant found in this study may lead to loss of function (LoF). According to the gnomAD database, this variant is absent in normal populations.
The protein structure of β-globin gene is shown in Figure 2. Full-length amino acid translation consisted of 174 amino acids. The β-globin gene consists of a nucleotide encoding a protein called beta-globin. β-globin is a component (subunit) of a larger protein called hemoglobin, which is located inside red blood cells. In adults, hemoglobin consists of four protein subunits: two subunits of beta-globin and two subunits of a protein called alpha-globin, which is produced from another gene called HBA (both HBA1 and HBA2). Each of these protein subunits is attached (bound) to an iron-containing molecule called heme, and the iron in the center of each heme can bind to one oxygen molecule. Hb within red blood cells binds to oxygen molecules in the lungs. These cells then travel through the bloodstream and deliver oxygen to the tissues throughout the body. A full-length protein will also have normal function, whereas a mutation in the gene will lead to disease.
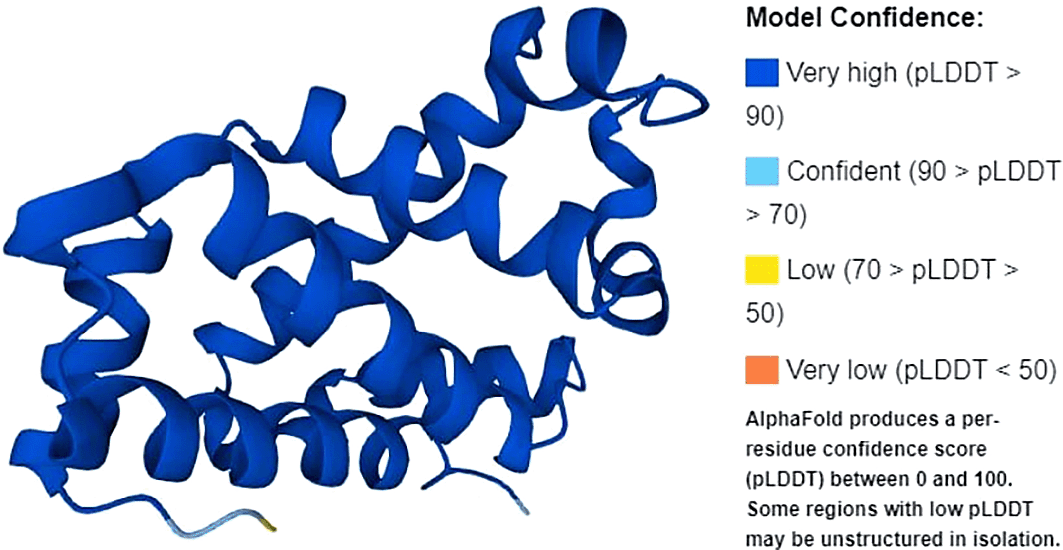
Source: Uniprot.
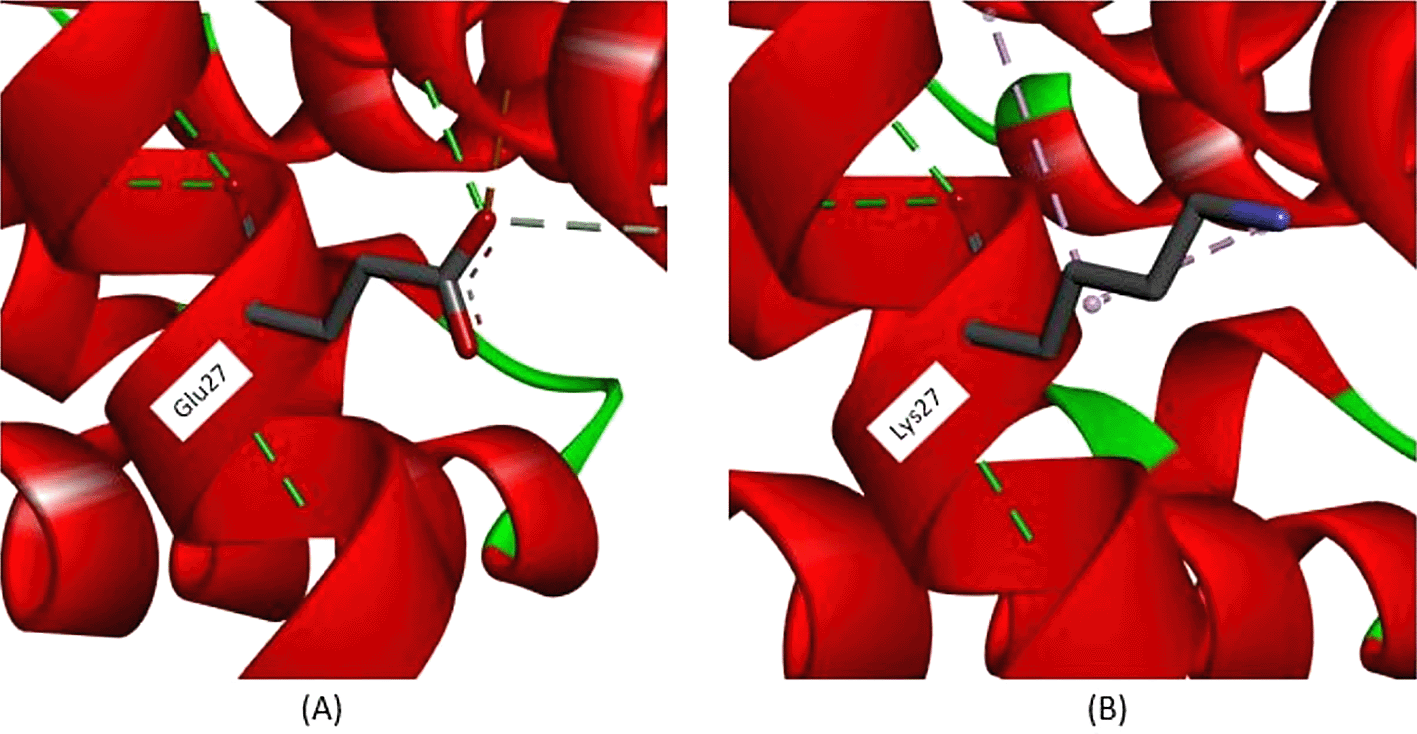
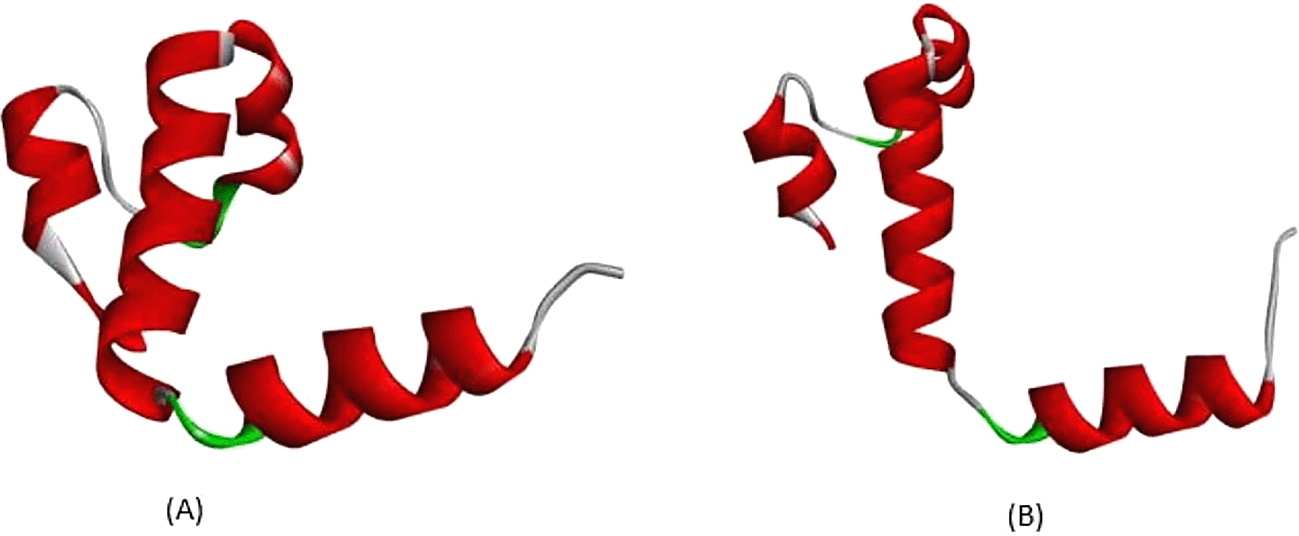
All exonic variants found in this study lead to amino acid changes that affect their interactions. According to Figure 3, a variant occurs in c.79, with nucleotide changes from guanine (G) to adenine (A), leading to an amino acid change at position 27. The amino acid change from glutamine (Glu) to lysine (Lys) is predicted to disrupt hemoglobin function. According to the analysis depicted in Figure 3A, in normal protein translation, glutamine at position 27 has a conventional hydrogen bond with Glycine30 (3.36 Å), Arginine31 (2.85 Å), and Glutamine23 (2.95 Å), as shown by the green dotted line. The carbon-hydrogen bond is also present between Glutamine27 and Histidine117 (3.55 Å), as indicated by the grey dotted line. An important interaction is the salt-bridge interaction between Glutamine27 and Arginine31 (2.71 Å), shown with a brown dotted line. Salt-bridge interactions are known to maintain this protein in equilibrium. While the variant Cd26 (HGVS: c.79G>A) led to a change from Glutamine27 to Lysine27, this structure is depicted in Figure 3B. Conventional hydrogen bonds remain; however, there are no salt-bridge interactions. The salt-bridge interaction changes to an alkyl interaction between Lysine27 and Arginine31 (5.31 Å) and Lysine27 and Valine114 (4.15 Å), as indicated by the pink dotted line. Loss of salt-bridge interactions causes an imbalance in protein structure.
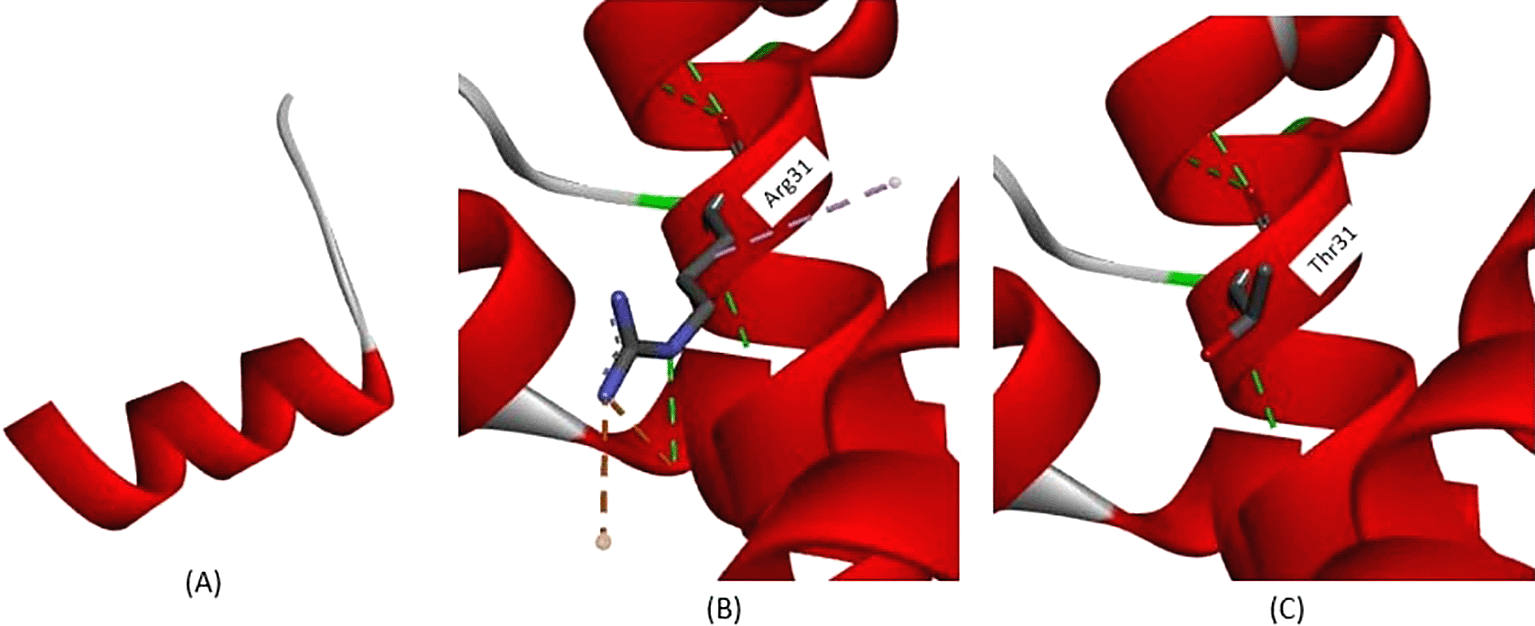
Variants occur at c.47, which is commonly known as the Cd15 mutation, and nucleotide changes from guanine (G) to adenine (A) lead to amino acid changes at position 16. This variant resulted in amino acid changes from Tryptophan to “terminated” amino acid, known as premature stop codon. This leads to incomplete protein translation because the final product has only 16 amino acids, whereas the normal protein translation from the β-globin gene has 147 amino acids. This amino acid change is depicted in Figure 4A, while the normal protein structure was depicted beforehand (Figure.2). Another variant observed in this study was a nonsense variant, Cd30 (HGVS: c.92G>A); the nucleotide changes from Guanine to Adenine lead to amino acid changes at position 31. According to the analysis depicted in Figure 4B, on normal protein translation, arginine at position 31 has a conventional hydrogen bond with Valine34 (3.37 Å), Valine35 (2.84 Å), and Glutamine27 (2.85 Å and 2.77 Å) also Arginine31, and Histidine117 (4.10 Å), shown with a green dotted line. The salt-bridge interaction between Arginine31 and Glutamine27 (2.71 Å) is indicated by a brown dotted line. An alkyl interaction was also observed between Arginine31 and Valine110 (4.98 Å). The variant Cd30 (HGVS: c.92G>A) leads to a change from Arginine31 to Threonine31, this structure is depicted in Figure 4C. The conventional hydrogen bond still remains; however, both salt-bridge and alkyl interactions are not observed due to amino acid changes. Loss of both salt bridges and alkyl interactions disrupts the overall protein structure and function.
One frameshift variant (common name Cd41/42 (HGVS: c.126_129delCTTT)) leads to amino acid changes from proline at position 42 to leucine, and a premature termination codon occurs after translation of 17 amino acids, as depicted in Figure 5A. This frameshift variant produced a shorter protein product, which is in accordance with the severity of the subject. A rare variant observed in this study was c.110delC, which led to an amino acid change from proline at position 37 to leucine, followed by a premature stop codon after 23 amino acid translations. According to the analysis depicted in Figure 5B, genetic variations shortened the end-protein product. This strengthens the prediction that frameshift variants greatly influence overall protein structure, interactions, and bonds. In addition, the subject's clinical condition matched all variants found in this study.
Thalassemia is the most common inherited hemoglobinopathy in Indonesia. Ten pathogenic variants were identified in this study. The variants observed consisted of intronic variants, missense variants, nonsense variants, and frameshift variants. The most common variant found was IVS1nt.5 (HGVS: c.92+5G>C; p.(?)) and Cd26 (HGVS: c.79G>A; p.(Glu27Lys)) variants. Functional in silico analysis was performed for each exonic variant, and the amino acid changes in the interactions and translations were explained.
This study was approved by the Ethics Committee of the Faculty of Medicine, Universitas Indonesia (No. KET-600/UN2.F1/ETIK/PPM.00.02/2023). All subjects recruited for this study signed an assent form.
All participant recruited for this study is already signed a written consent form. The consent form has been explained verbally to all the participant before the signage. All participant already understands the research and all the content included within the written consent form.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)