Keywords
functional study, glycogen branching enzyme, Indonesia, novel mutation, rare disease
functional study, glycogen branching enzyme, Indonesia, novel mutation, rare disease
Glycogen Storage Disease IV/Andersen disease (GSD IV [MIM: 232500]) is a functional rare disease (RD) that causes abnormalities in glycogen synthesis and degradation.1 GSD IV is caused by a defect in the GBE1 gene [MIM: 607839], which encodes the 1,4-alpha-glucan-branching enzyme (GBE). GSD IV occurs in 1 every 760,000–960,000 live births and increases in regions that have numerous consanguineous marriages.1,2 Deficiency of functioning GBE results in the deposition of amylopectin-like polyglucosan, which is deposited in the liver, muscle, cardiac muscle, bone marrow, central nervous system, and peripheral nervous system.3 Classic natural history of GSD IV is hepatosplenomegaly and failure to thrive at 18 months of age, with hypoglycemia and progressive liver failure resulting in mortality before the age of 5 years.3,4 Liver transplant is still the recommended first-line therapy for early GSD IV with isolated hepatic abnormality. To prevent the progression of the disease, dietary modifications such as fasting avoidance, increasing protein intake, uncooked cornstarch staples, and continuous nocturnal gastric feeding aimed to retain euglycemic conditions.4 Among pathogenic GBE1 variants causing GSD IV, GBE1 L224P, and Y329S variants are the most studied variants due to their prevalence among GBE1 missense mutations. The GBE1 L224P variant represents a near-zero GBE level, leading to severe progressive clinical manifestation with a high mortality rate, while the Y329S variant represents a partial decrease in GBE level with non-progressive clinical manifestation.5–8
Diagnosing RD for healthcare providers remains a challenge due to the atypical clinical presentation and different natural history of RD in each patient. This challenge is further exacerbated by a lack of national policies, financial support, referral facilities, genetic experts, and clinical guidelines in developing countries.9 Diagnosing RD usually takes around 5.6–7.6 years with poor prognosis and life-threatening complications,10 and may require single omic to multiomics study11–13 to establish its pathogenicity according to the American College of Medical Genetics and Genomics (ACMG) classification.14–16 Here, we report a novel missense mutation in GBE1 R198T, inherited from heterozygous parents, which proved to be pathogenic after combined multiomics in silico and in vitro functional studies. We further investigated the pathomechanism GBE1 R198T by molecular modeling of this protein and compared it with well-known mutations, such as L224P and Y329S. We also discuss the combined clinical and gene-phenotype analysis with further protein structural prediction to help functional RD diagnosis in developed countries. To our knowledge, this is the first study on the pathogenicity establishment of GBE1 R198T novel variants, and the first comprehensive rare disease multiomics with functional study in Indonesia.
Clinical information was obtained from the attending physician from 09 to 31, 2023. All meaningful findings were coded to the standard terminology HPO before being analyzed using the in-house phenotype-genotype pipeline IDeRare.17
Cryopreserved DNA samples were isolated from blood samples taken from the patient and the patient’s parents using the Geneaid™ DNA Isolation Kit (Blood) according to the manufacturer’s protocol on May 20, 2021. Sequencing was conducted at the Beijing Genomics Institute (BGI) Genomics Laboratory, China, using DNA Nanoball Sequencing (DNBSeq) and Agilent V6 kit with 100x coverage on March 26, 2023, with Material Transfer Agreement (MTA) approved by the Ministry of Health Indonesia (HK.07.01/H/2443/2023). The resulting sequencing files in FASTQ format were analyzed, annotated, screened, and prioritized using the IDeRare pipeline.17 Novel VUS was defined as rare variants with minor allele frequency < 0.05 with a lack of population statistics information or functional effects of mutations in the population. Population databases used to confirm the novelty of this variant were the gnomAD3.1 database18 (https://gnomad.broadinstitute.org/) and dbSNP.19 Functional prediction of the mutations was performed using the dbNSFP 4.4a20 annotation database.
In this study, the wild-type (WT) GBE sequence was obtained from NCBI (GenPept #NP_000149.4) and edited to create novel R198T, known pathogenic L224P, and Y329S amino acid sequences. WT, L224P, Y329S, and R198T were modeled using AlphaFold2,21 validated with MolProbity,22 and superimposed using PyMol.23–25 Structural analysis of mutations was compared to the normal GBE protein residue domain, which was grouped into the N-terminal alpha helix (43–75), carbohydrate binding molecule 48 (CBM48, 76–183), central catalytic core (184–600), and C-terminal amylase-like domain (601–702) with a ligand binding site involving surface proteins from both CBM48 and the central catalytic core (62–63, 91–93, 118–121, 333–335).6,26
Identification of potential changes in the primary structure (chemical bonding, hydrophobicity, and steric hindrances), secondary structure (protein misfolding), and tertiary structure (change in protein surface) between novel R198T mutations and normal (WT) or known-pathogenic mutations (L224P, Y329S) was performed using ExPASy,27 PSIPRED,28 and visualized using PyMol. Prediction analysis of proteasome degradation through internal and external proteasome systems was performed using the proteasome cleavage prediction server (PCPS),29 with the ubiquitination tendency of lysine (Lys, K) in the ubiquitin-proteasome system (UPS) pathway30 analyzed using RUBI.31
Maltoheptose (Glc7) [Glygen #G41288IQ]6 was used to model the binding site of the glucose polymer in the GBE. Potential binding sites and cavities were predicted using the ICMPocketFinder. Protein-ligand docking was performed using Autodock Vina with grid coordinate centers Cx, Cy, Cz (−13.514, 7.876, 27.993) and box dimensions of sx, sy, sz (40, 40,40), centered near the ligand-binding site of normal GBE (GBE WT). The highest-rank complexes with the lowest Gibbs free energy were visualized using the BIOVIA Discovery Studio Visualizer.
HEK293T cells were grown in Dulbeccos’s modified Eagle’s medium (Gibco) with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin at 37 °C with 5% CO2. The cells were transiently transfected with plasmids from Synbio Technologies (USA) encoding eGFP-vector/empty vector (EV), WT, L224P, Y329S, and R198T using Lipofectamine™ 3000 Transfection Reagent (Invitrogen™ # L3000008) according to the manufacturer’s protocol. The cells were grown overnight (20 h) in basal medium before harvesting. The expression of β-actin and GBE from triplicate-independent experiments was analyzed by western blotting.
The cells were lysed in RIPA buffer supplemented with a protease inhibitor cocktail (Roche #05892970001) at a ratio of 5 × 106 cells/1 mL of buffer. The lysis process was performed on ice for 15 min before being centrifuged at 12,000 rpm for 5 min at 4 °C. The supernatant was collected and quantified using a DC Protein Assay (Biorad #500–0116) and read on an ELISA reader at a wavelength of 655 nm. Equal mass of protein (10 μg) supernatant was boiled in Laemmli Sample Buffer for 5 min. The samples were separated using SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Merck Millipore #IPVH00010) at 100 V for 1 h. The membranes were blocked in 5% milk (BIORAD #1706404) in TBS-Tween for 1 h before being incubated with 1:1000 primary antibodies rabbit anti-GBE (Thermo #PA5–26515) and 1:2500 rabbit anti-β-actin (Thermo #PA5–85271) overnight at 4 °C. The membranes were then washed three times with TBST, labeled with 1:10000 horseradish peroxidase (HRP)-conjugated secondary antibodies goat anti-rabbit (Thermo #31460), and detected by enhanced chemiluminescence (ECL) (Elabscience #E-IR-R307) using ImageQuant™ LAS 4000. The band signal was analyzed using ImageJ, and the GBE (~72 kDa) signal was normalized to the β-actin (~42 kDa) signal.
GraphPad Prism 9 was used for the analysis of in vitro results. The normality test was done using the Shapiro-Wilk test, and Analysis of Variance (ANOVA) was used to compare band intensities between mutants and wild type. Post hoc analysis was conducted using Tukey’s multiple comparisons for multiple comparisons between each group. Statistical significance was defined as P < 0.05.
Patient A was a 3-year-9-month-old Javanese boy referred from a regional referral hospital, Dr. Kariadi Hospital to the Nutrition and Metabolic Department of Child Health, Dr. Cipto Mangunkusumo National Referral Hospital (RSCM), Jakarta on March 24, 2021 because of hepatosplenomegaly since the age of 2 years with a history of weight faltering, refractory anemia, and generalized weakness. He was second-born to an asymptomatic non-consanguineous 37-year-old father and 32-year-old mother. The patient was delivered full-term by C-section with normal birth weight and length. His elder brother passed away at 2 years of age with an enlarged abdomen, which was the cause of death due to suspected thalassemia. His maternal aunts were known to have unexplained death before the age of 5 years (Figure 1). Before his visit, he had consumed ursodeoxycholic acid (2 × 75 mg), folic acid (1 × 1 mg), and albumin (3 × 1000 mg) daily.
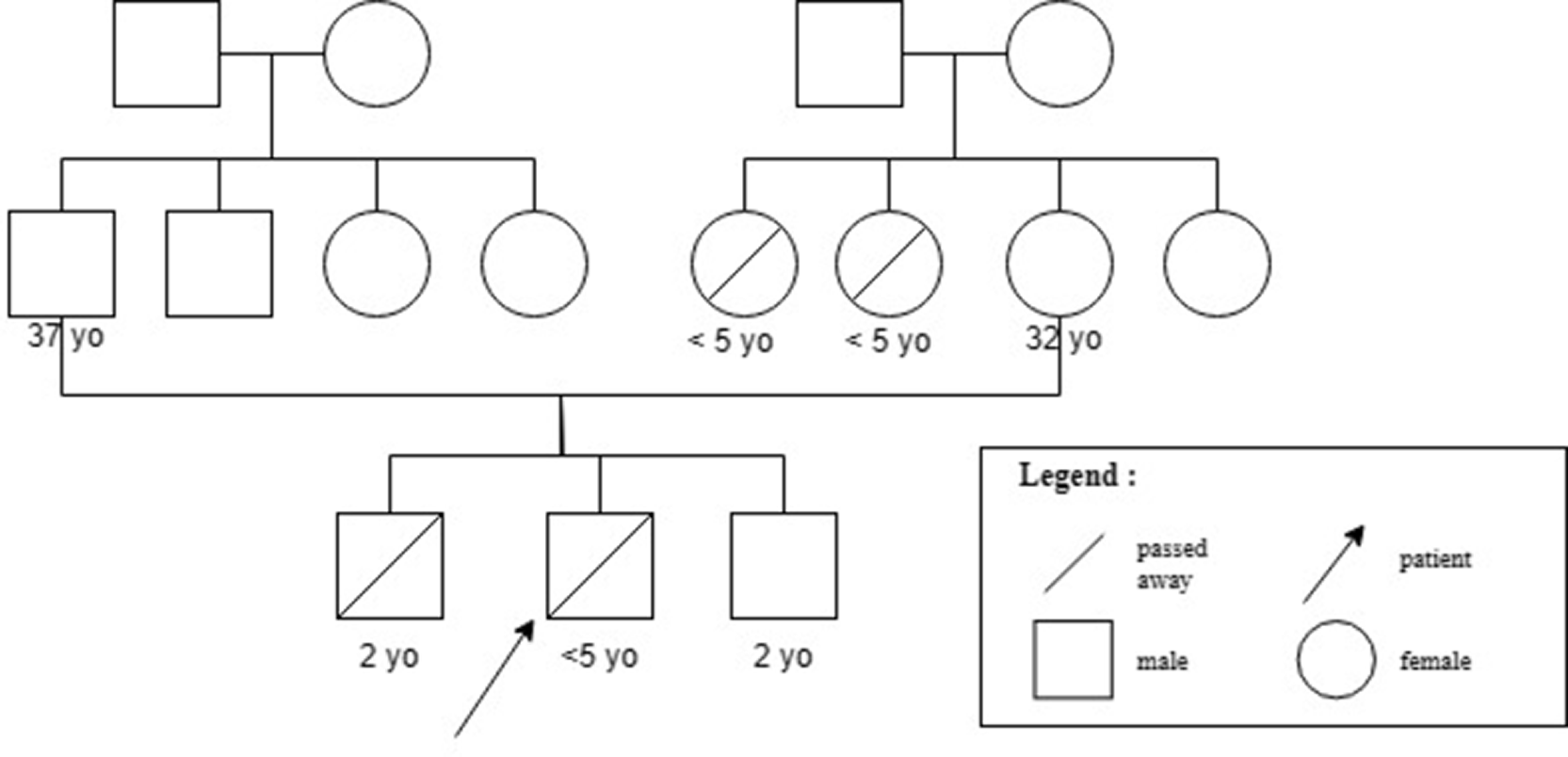
Family pedigree of Patient A (black arrow) harboring GBE1 R198T.
Physical examination at the clinic revealed anemic conjunctiva, enlarged liver 4 cm below the costal arch, spleen in the Schuffner 4 position, and enlarged abdominal circumference of 57 cm with no specific dysmorphism. Anthropometric examination showed mid upper stature (14.3 cm, Z-score − 2 to −1 SD), and normal stature (94.5 cm, Z-score − 2 to −1 SD). Laboratory workup showed anemia (10.5 g/dL), thrombocytopenia (79 x 103/μL), increased transaminase enzyme (ALT 136 U/L, AST 308 U/L), GGT of 82 U/L, cholestasis (total|direct|indirect bilirubin 3.17|1.74|1.43 mg/dL), hypoalbuminemia (3.13 g/dL), PT/APTT: 1.46x/1.5x, and normal fasting blood glucose (68 mg/dL). Follow-up blood metabolomic result: mild hyperlactatemia (3.0 mmol/L), AFP 26.09 ng/mL, metabolic alkalosis (pH 7.541, pCO2 29.6 mmHg, pO2 51 mmHg, HCO3 25.6 mmol/L, Total CO2 26.5 mmol/L, BE 4.2 mmol/L, O2 saturation 89.7%), mild hyponatremia (sodium 132 mEq/L, potassium 3.9 mEq/L, chloride 102.8 mEq/L, calcium 8.1 mg/dL, calcium ion 1.17 mmol/L, phosphate 4.3 mg/dL, magnesium 1.73 mg/dL), normal ammonia (85 μg/dL), and low HDL (Total cholesterol 122 mg/dL, HDL 26 mg/dL, LDL 85 mg/dL, Triglyceride 67 mg/dL).
Before referral, the patient was treated in the hematooncology department of a regional hospital from his first admission at the age of 2 years, before finally being referred to the nutrition and metabolic department at 3-year-2-month-old. The patient underwent numerous initial and follow-up diagnostic procedures in regional hospitals, including routine hematological, chemical, and immunological workup; hemoglobin electrophoresis; bone marrow biopsy; abdominal US and MSCT; bone survey; echocardiography; liver biopsy; hepatobiliary scan; and electromyography. Liver biopsy showed a red purplish color at hepatocyte cytoplasm inclusions on periodic acid-Schiff (PAS), with progression of liver cirrhosis. Bone marrow biopsy showed the existence of foam cells supporting lysosomal storage disease. A nerve conduction study showed a proximal lower motor neuron disease origin. Bone scans only showed osteopenia, echocardiography showed mild mitral regurgitation, abdominal US and MSCT supported hepatosplenomegaly, and hepatobiliary scans excluded biliary atresia. Several differential diagnoses were considered and ruled out upon diagnostic workup, including thalassemia, autoimmune hepatitis, common cause of liver injury or infective hepatitis, hepatoblastoma, biliary atresia, myelodysplastic syndrome, Gaucher disease, and Niemann Pick type C. Proband genetic screening was conducted after the exclusion of all potential differential diagnoses when the patient was 3-years and 5-months-old. All notable clinical findings were coded in HPO (Supplementary Table 1).
A novel GBE1mutation (NM_000158.4:c.593G > C:p. R198T) was identified by proband and trio sequencing of the heterozygous parent (ACMG PP4, Figure 2A). Variant-based prioritization conducted using IDeRare17 detected no other known pathogenic mutations in GBE1. The frequency of this variant in the population has not been recorded in gnomAD3.1(+) or reported in NCBI dbSNP (ACMG PM2, Table 1). GBE1 R198 is a conserved region of GBE1. Annotation analysis using SIFT4G, PolyPhen2, MutationTaster, PROVEAN, MetaSVM, MetaLR, MetaRNN, and MVP consistently supported the damaging nature of the R198T mutation ( Table 1). The REVEL ensemble score of 0.873 supported moderate pathogenicity16 in silico (ACMG PP3, Table 1). In addition, based on August 09, 2024, variant data, most missense GBE1 variants (44 out of 50) were classified as pathogenic or likely pathogenic (ACMG PP2).32
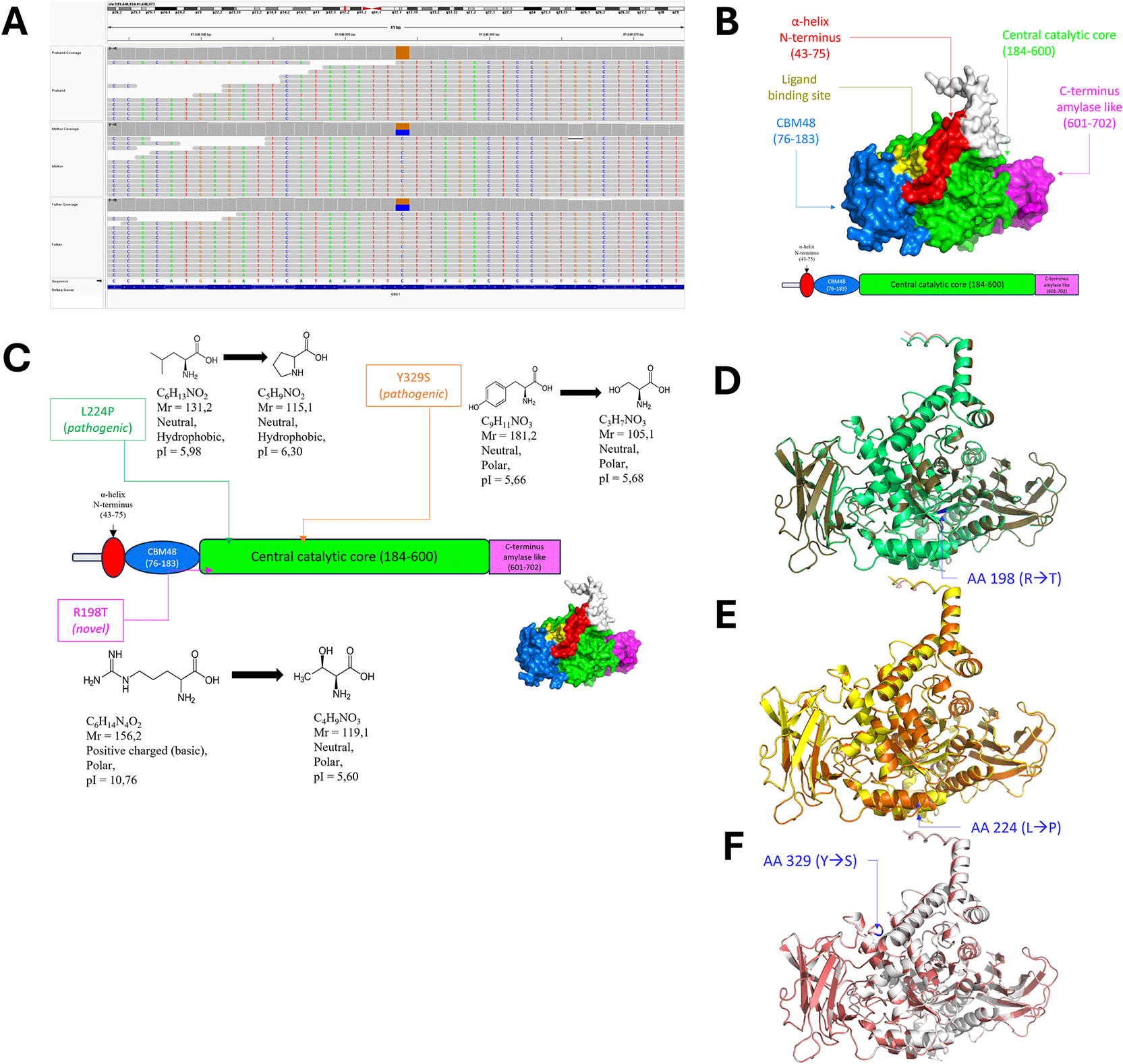
(A) Visualization of GBE NM_000158.4:c.593G > C:p. R198T of the proband and parents. (B) 3D model of GBE WT grouped by the color of its domain red: N-terminal alpha helix; blue: CBM48; green: central catalytic core; pink: C-terminus amylase-like structure. (C) Amino acid changes in R198T, L224P, and Y329S within the GBE domain. (D-F) Superimposition of WT (red) with R198T (D, light green), L224P (E, yellow), and Y329S (F, white).
| Parameter | L224P | Y329S | R198T |
|---|---|---|---|
| Coordinate location (chr-loc) | 3–81648876 | 3–81642787 | 3–81648954 |
| Codon mutation | A > G | T > G | C > G |
| Amino acid mutation | L > P | Y > S | R > T |
| dbSNP rsid | rs137852886 | rs80338671 | N/A |
| ClinVar & Klasifikasi ACMG | Pathogenic | Pathogenic | Unknown |
| SIFT4G | 0.92824 (D) | 0.92842 (D) | 0.48855 (D) |
| Polyphen2 | 0.92359 (D) | 0.92359 (D) | 0.92359 (D) |
| MutationTaster | 0.81001 (A) | 0.58761 (D) | 0.54805 (D) |
| FATHMM | 0.93835 (D) | 0.86963 (D) | 0.85799 (D) |
| PROVEAN | 0.89759 (D) | 0.97155 (D) | 0.87143 (D) |
| VEST4 | 0.99548 (D) | 0.85979 (D) | 0.89800 (D) |
| MetaSVM | 0.97522 (D) | 0.97025 (D) | 0.95207 (D) |
| MetaLR | 0.96872 (D) | 0.95387 (D) | 0.94659 (D) |
| MetaRNN | 0.95005 (D) | 0.28169 (T) | 0.87921 (D) |
| GERP* | 5.12 | 6.06 | 5.92 |
| REVEL** | 0.946 | 0.979 | 0.873 |
| MVP | 0.93518 (D) | 0.93231 (D) | 0.98481 (D) |
| gnomAD3.1 (allele frequency) | 1/50,000 | 1/5,000 | #N/A |
| • Ashkenazi Jewish | 0.0005764 | 0.006928 | #N/A |
| • European (non-Finnish) | 0.00001472 | 0.00007355 | #N/A |
| • XX | 0.00001285 | 0.0002444 | #N/A |
| • XY | 0.00002692 | 0.0001347 | #N/A |
| Total | 0.0001972 | 0.001908 | #N/A |
Molecular modeling was performed to explore the effect of the R198T mutation on the overall structure of the novel GBE1 R198T mutant compared with normal GBE1WT, pathogenic L224P, and Y329S mutants. Base substitution (G replaced by C) resulted in a coding change from arginine (Arg, R) to threonine (Thr, T) at the 198th amino acid residue. The R198T mutation is located in the central catalytic core domain of GBE, which plays an important role in catalyzing glycogen branch formation, similar to L224P and Y329S (Figure 2C).
The AlphaFold models of GBE1 R198T, L224P, Y329S, and WT were validated and superimposed. The overall modeled structure was well-modeled, with Ramachandran favoring ≥95% and MolProbity Score ≥ 66th percentile (Supplementary Table 2), with Ramachandran Plot attached in Supplementary Figure 1–3. The superimposition RMSD values of all mutants were < 2 Å, indicating minimal deviation of the overall structure of the SNV mutant compared to that of the GBE WT (Figure 2D-F, Supplementary Table 2).
The significant change in the isoelectric point (pI) of GBE1 R198T was caused by the loss of positively charged arginine, which was replaced by threonine (neutral charge). GBE1 Y329S experienced a greater reduction in molecular weight than the other mutants due to the loss of the tyrosine phenol ring. The aliphatic index of GBE1 L224P was decreased by the loss of leucine, which has an aliphatic side chain, and its replacement with proline, which has a cyclic side chain. The Y329S mutation, compared to other mutants, exhibited hydrophobicity properties that were close to those of GBE1 WT (GRAVY Y32S -0.374 vs. WT −0.375), reflecting the preserved hydrophobicity of the GBE1 Y329S protein. All GBE mutants were categorized as stable in vitro (Supplementary Table 3).
The results of the internal proteasome and immunoproteasome cleavage site analysis showed R198T degradation change(s) at the 198th residue, L224P at the 223rd and 224th, and Y329S at the 328th and 329th (Supplementary Table 4). The ubiquitination prediction results showed a slight decrease and increase in the ubiquitination tendency of R198T and L224P, respectively, compared to WT. Y329S had the same ubiquitination probability as WT, indicating a relatively similar ubiquitination pattern between Y329S and WT (Supplementary Table 5).
The GBE1 R198T mutation led to the disruption of polar contacts and loss of steric hindrance formed by G231 and E591. The polar contact with C234 is shortened (from 2.5 Å to 2.4 Å). Å N233 increased from two contact points (2.3 Å and 2.1 Å) to three contact points (2.3 Å, 2.1 Å, 2.3 Å), as shown in Figure 3A-B. The disruption of the bond with G231 and E591 caused disarrangement of the GBE catalytic core protein domain with the alpha helix, CBM, and ligand-binding site (Figure 4A) and secondary structure misfolding (Supplementary Table 6) due to the loss of the strong bond between arginine and negatively charged amino acids such as E591. The short side chain of T198 prevented it from bonding with G231, and stronger polar contacts with C234 and N233 changed the protein surface curvature near T198 (Figure 4G-H).
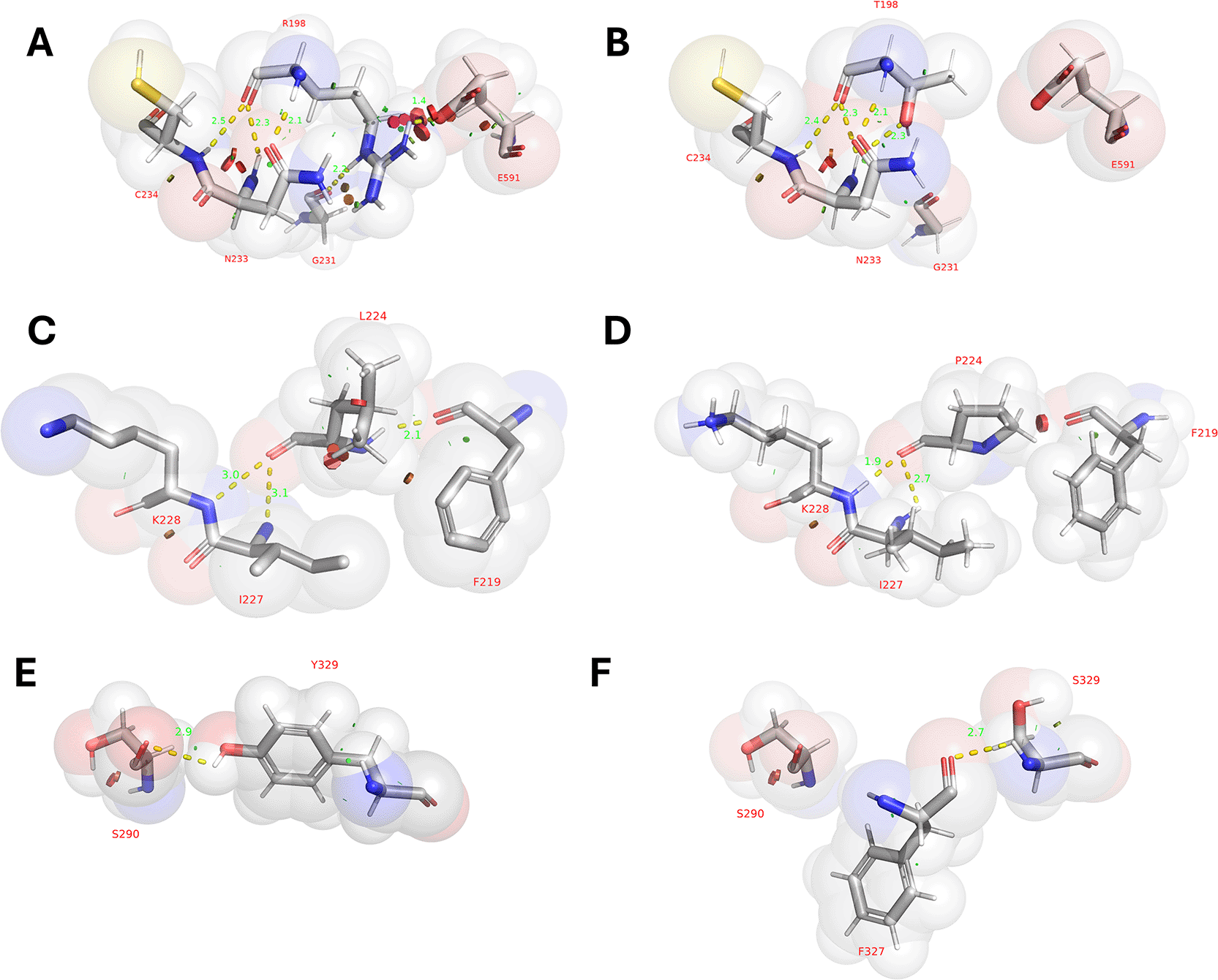
Red bump = steric hindrance.
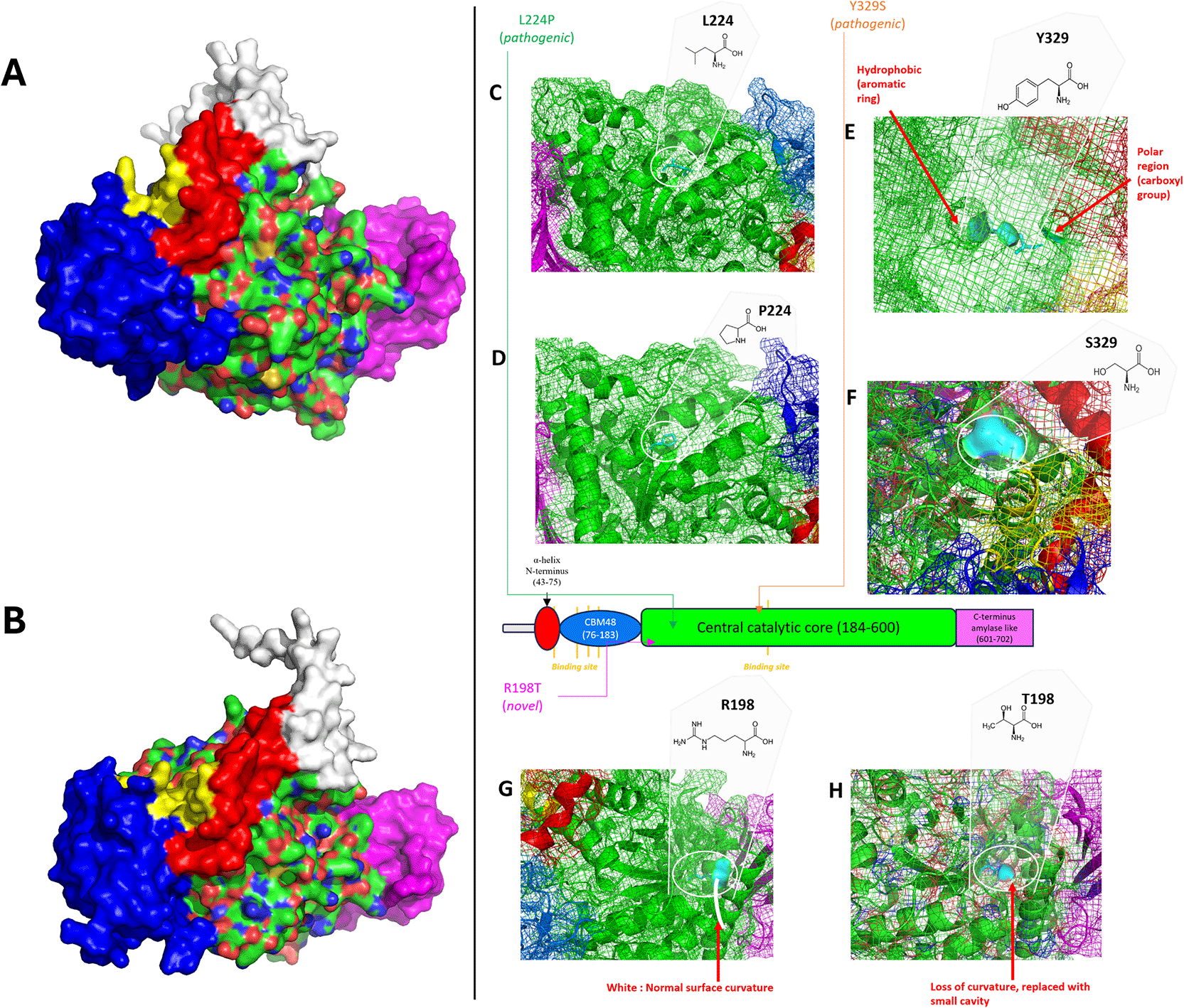
GBE domain surface disarrangement in (A) GBE1 R198T and (B) Y329S. Comparison of the normal protein surface of GBE1 WT: (C) L224, (E) Y329, (G) R198, compared to changes caused by (D) L224P, (F) Y329S, (H) R198T mutations; red: N-terminal alpha helix, blue: CBM48, green: central catalytic core, pink: C-terminus amylase-like structure, yellow: amino acid involved ligand binding site in normal GBE, cyan: mutation hotspot.
Similarly, the GBE1 Y329S mutation caused a loss of the hydrophobic phenol core, the disruption of polar contact with S290, and the formation of a new bond with F327 (2.7 Å) (Figure 3E-F), resulting in disarranged protein domain (Figure 4B) and replacement of hydrophobic and polar buried cavities into single protrusion polar cavity (Figure 4E-F). Meanwhile, GBE1 L224P mutation disrupted polar contacts and increased steric hindrance with F219 due to the cyclic ring of P224 (Figure 3E-F). Under normal conditions (WT), L224 was not an amino acid located beneath the surface (Figure 4C-D) and the L224P mutation did not cause the disarrangement of the GBE protein domain because the existence of polar bonds with F219, I227, and K228 was retained (Figure 3C-D).
The docking predictions were validated by comparing the rank 1 docking conformation of GBE1 WT with a previous study conducted by Froese et al. (2015). The docking results showed Glc7 contact GBE1 WT at amino acids 62–63, 91–93, 111–122, 332, and 336, similar to the findings reported by Froese et al. (2015) (Fig. S5). GBE1 L224P (Fig. S6) and R198T (Figure 5) mutations caused changes in the Glc7 ligand-binding sites. GBE1 Y329S (Fig. S7) mutants had relatively the same ligand-binding site as GBE1 WT involving amino acids both in the CBM48 domain (62–63, 68–69, 71–72, 76, 85, 89, 91, 93–94, 116, 119–121) and central catalytic core domain (332–333, 336) despite its disarranged surface (Figure 4B).
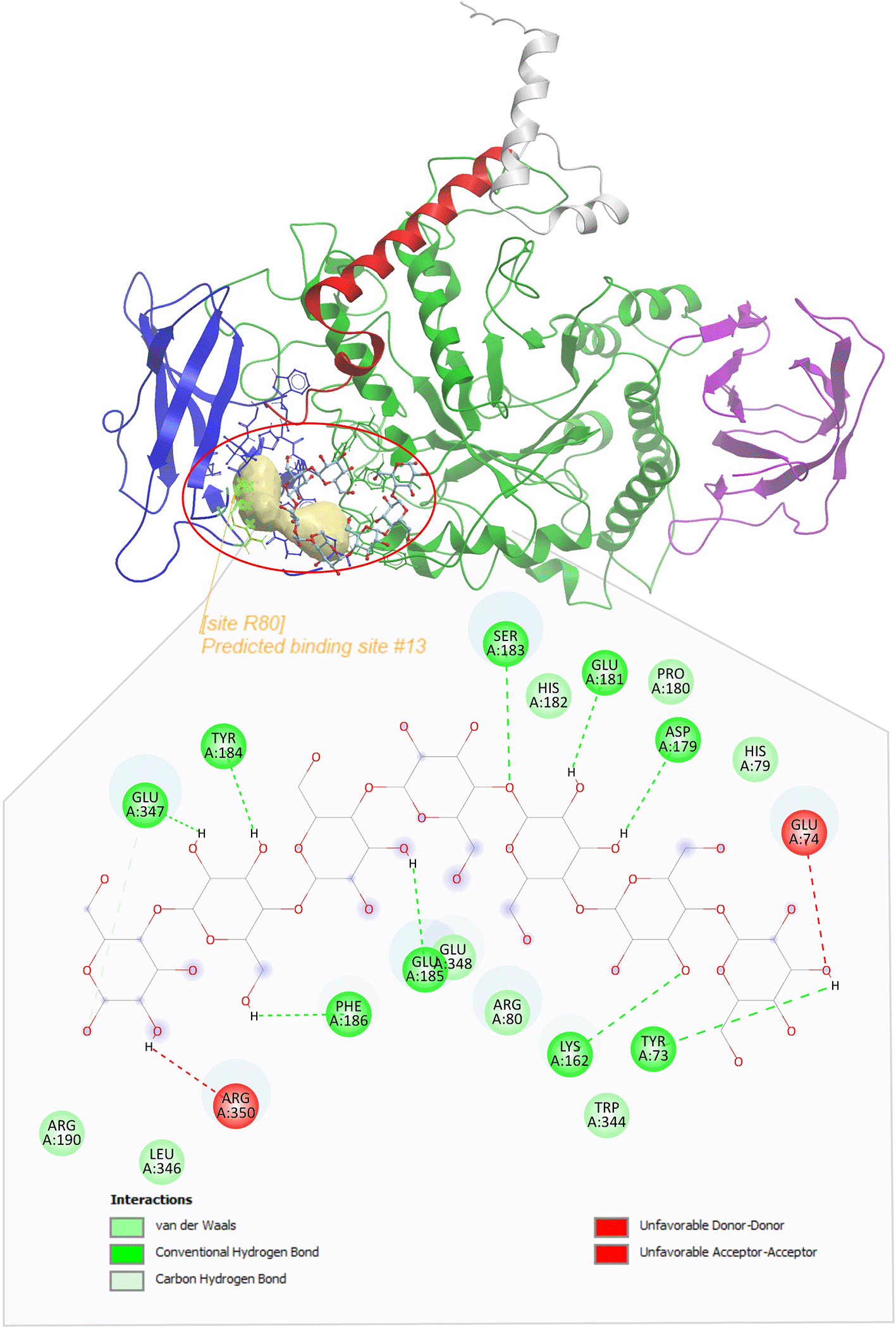
Protein ribbon: Catalytic core (green), alpha helices (red), CBM (blue), and C-terminus amylase-like (magenta). The predicted binding site for the GBE1 R198T-Glc7 ligand is located around R80 (red circle), with a pale yellow cavity. The GBE1 R198T -Glc7 ligand binds through hydrogen bonds with TYR73, LYS162, ASP179, GLU181, SER183, TYR184, GLU185, PHE186, and GLU347, with weak non-covalent carbon-hydrogen interactions on HIS79, ARG80, PRO180, HIS182, ARG190, TRP344, LEU346, and GLU348, and donor-donor interactions on GLU74 and ARG350.
The predicted binding site for the GBE1 R198T-Glc7 ligand (Figure 5) is located around R80 (red circle). The GBE1 R198T-Glc7 ligand binds through hydrogen bonds with TYR73, LYS162, ASP179, GLU181, SER183, TYR184, GLU185, PHE186, and GLU347, with weak non-covalent carbon-hydrogen interactions on HIS79, ARG80, PRO180, HIS182, ARG190, TRP344, LEU346, and GLU348, and donor-donor interactions on GLU74 and ARG350. Both L224P and R198T exhibited changes in ligand-binding sites located closer to the catalytic core, with the Glc7 ligand conforming in a circular manner on the cavity wall, in contrast to WT and Y329S, which adopt relatively ligand-receptor binding. The complete information on the available cavities on the R198T, L224P, and Y329S surfaces and protein-ligand Gibbs energies is presented in Supplementary Table 7.
Based on visual observations of the treatment group (Figure 6A), the β-actin band appeared relatively consistent across the columns, indicating a balanced load of protein across each lane. The GBE band showed lower signal intensity for the GBE1 R198T variant than GBE1 WT, indicating that protein expression had decreased. When compared to previously known pathogenic variants, GBE1 R198T protein expression was stronger than GBE1 L224P, but weaker than GBE1 Y329S. Normalization of GBE signals resulted in band intensity of GBE1 WT: 124880 ± 53946, L224P: 10406 ± 3092, Y329S: 24903 ± 11855, and R198T: 13878 ± 7605, respectively. The Shapiro-Wilk test showed normality across three independent experimental sets (P > 0.05), and the ANOVA test resulted in P = 0.0029. Subsequent multiple comparison tests across groups showed that GBE1 WT signal was significantly higher than all mutants: R198T (P = 0.0054), L224P (P = 0.0045), and Y329S (P = 0.0100) (Figure 6B, Supplementary Table 8), which supported the ACMG PS3 criteria for the R198T novel mutation. However, there were no significant differences between the R198T vs. L224P mutants (P = 0.9986), R198T vs. Y329S (P = 0.9606), or L224P vs. Y329S (P = 0.9175) (Supplementary Table 6).
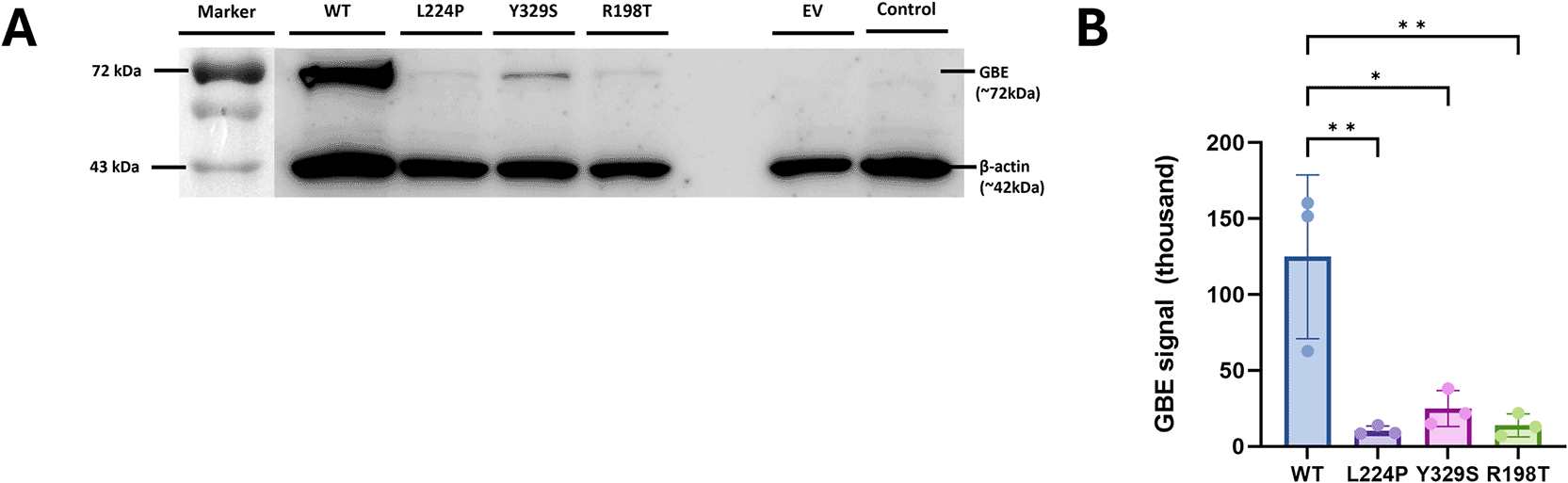
(A) Western blot analysis of HEK293T cells transfected with GBE1 wild type (WT), L224P, Y329S, R198T, empty vector (EV), and control (untransfected). (B) The GBE signal intensity across group; data are presented as mean ± SE, * indicates result P < 0.05, and ** indicates result P < 0.01 from one-way ANOVA.
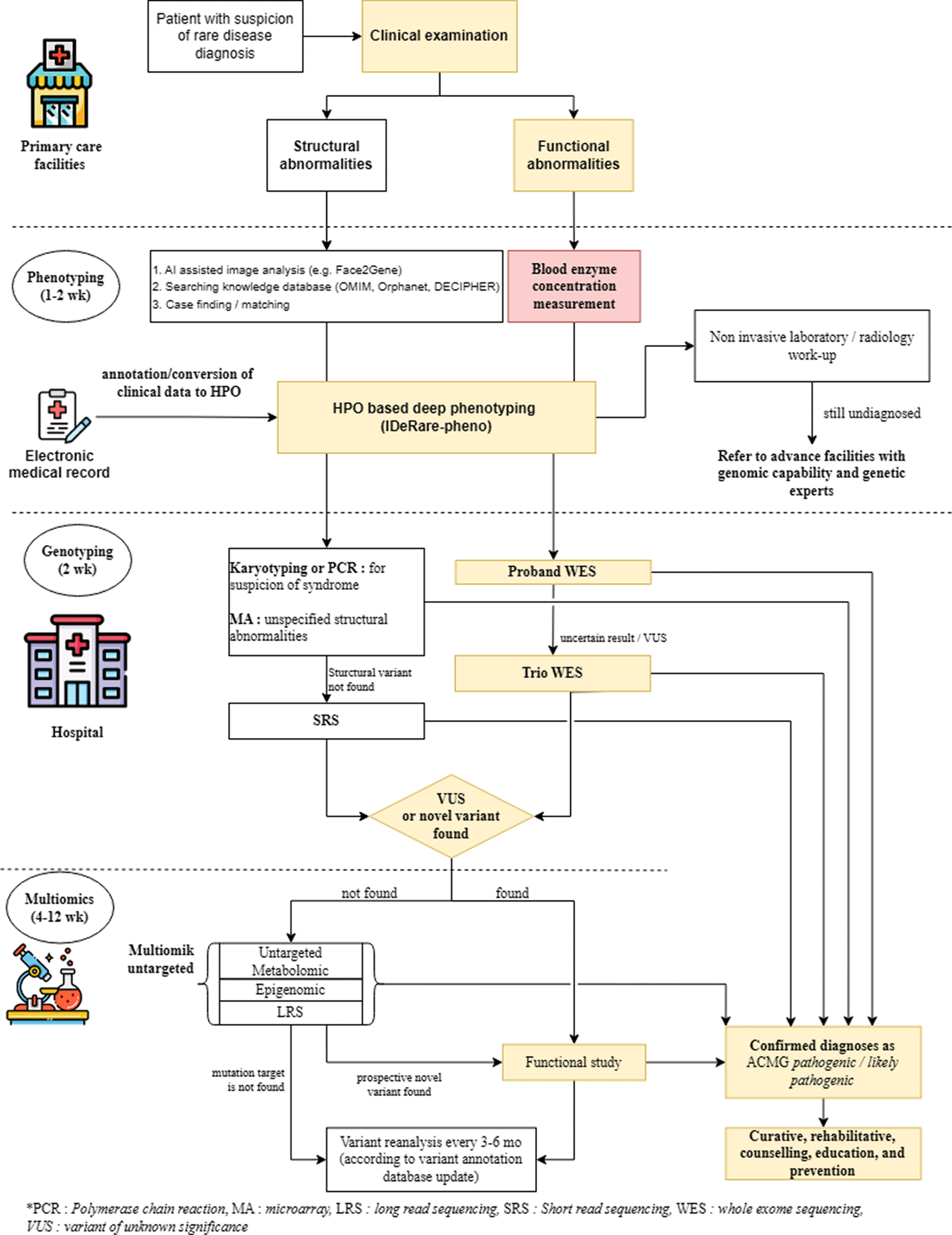
The yellow box indicates the steps that were conducted in this study; the red box was skipped because access to omics modalities was unavailable.
While phenotype-genotype analysis was able to match GBE1 R198T as a potential disease-causing variant, it was still classified as a VUS because this mutation only fulfilled one moderate and three supporting criteria (PM2, PP2, PP3, and PP4). The ACMG pathogenic mutation must be established by at least two moderate pathogenic and two supporting criteria (2 PM and ≥ 2 PP) or one strong pathogenic with two supporting criteria (1 PS and ≥ 2 PP criteria). Due to a lack of fulfilling moderate criteria, a single strong pathogenic criterion, such as PS3, was chosen. The protein expression study showed a lower GBE1 R198T novel variant band intensity compared to the GBE1 WT, fulfilling the PS3 criterion, which allowed us to reclassify the GBE1 R198T variant as likely pathogenic (1 PS criterion and ≥ 2 PP criteria).
Our multi-omics diagnostic experiment for GSD IV successfully demonstrated the pathogenicity and pathomechanism of GBE1 R198T. The experimental workflow was conducted step-by-step, starting with phenotype analysis, followed by genotype analysis and protein expression functional studies. As there is a lack of multiomics modalities in Indonesia and possibly in other developing countries, we proposed a modification of multiomics diagnostic workflows by prioritizing phenotype and genotype analysis before advanced omics studies. The modified workflow emphasized the importance of completing phenotype information and basic laboratory or radiological work-up within 1–2 weeks, before deciding the need for genotype analysis. Given the chance of encountering novel variants in rare disease cases, broad variant screening through WES was preferred over WGS, considering cost efficiency. Trio WES was proposed to identify patients’ VUS inheritance patterns and enhance the variant calling results. Genotype analysis can be completed within two weeks, considering that WES typically takes 5–10 days, with proband and trio analysis using the IDeRare pipeline requiring another day (~18 h).
Here, we report the identification of a novel GBE1 R198T mutation previously classified as VUS. This novel mutation has not been previously reported in the population allele frequency databases, gnomAD or dbSNP. The patient exhibited classical clinical symptoms of hepatic GSD and hepatosplenomegaly, which were masked as having a hematologic origin. Late consideration of GSD as a differential diagnosis was understandable because there is overlapping hematological, hepatobiliary, or oncological disease origin with more prevalent diseases in Indonesia, such as thalassemia, biliary atresia, hepatitis, or hepatoblastoma. The diagnosis of GSD IV is usually performed via extensive genetic screening to rule out potential pathogenic mutants. While not specific for GSD IV diagnosis, pathognomonic findings including diastase-resistant red purplish hepatocyte cytoplasmic inclusion on PAS staining may help guide and narrow the diagnostic possibilities.3,33
Mutation analysis utilizing the IDeRare17 phenotype-based variant prioritization pipeline ranked GBE1 R198T as the sole causative variant, which was inherited from the carrier parent and was consistent with GSD IV inherited in an autosomal recessive manner. It is worth noting that the R198T mutation is conserved and is located in the central catalytic core domain. This location followed the trend where most pathogenic missense mutations were located in this domain.8 This was supported by the fact that the CBM48 domain of GBE plays an important role in binding with the glucose chain (ligand), and the catalytic core is the part that actively catalyzes the branching of glycogen.6 However, as there were no sufficient (likely) pathogenic ACMG criteria fulfilled yet, other integrative multiomics studies beyond phenotype and mutation analysis are necessary. We chose protein expression instead of other approaches because of the lack of previously reported pathogenic missense mutations at the GBE 198th residue, the lack of a specific GBE proteomic assay available in Southeast Asia, and the feasibility of laboratory infrastructure personnel available.
We chose L224P and Y329S as pathogenic comparisons for our novel mutant, as these are two of the most well-studied mutants, both in vitro5–8 and molecularly modeled6 as pathogenic controls. Protein function prediction using the dbNSFP annotation database confirmed the potential pathogenic nature of the R198T mutant. Primary structural analysis supported that there is a change and broken hydrogen bond with nearby residues, change in steric hindrance, change in chemical parameters (charge, side chain, and pI), change in cleavage site, and ubiquitination tendency caused by the replacement of arginine with threonine. The AlphaFold 3D molecular model successfully showed a change in secondary structure folding, a disarranged protein surface on the supposedly central catalytic core site, and an altered surface contour and cavity near residue 198. This change correlates with the ligand-binding site of R198T, which is displaced far from normal WT in the middle of the central catalytic core domain and conformed in a circular manner on the cavity wall, which is similar to L224P. Altogether, R198T underwent changes in the primary structure (hydrogen bond break, change of polar contact distance, steric hindrance), secondary structure, tertiary structure (surface, cavity, and hydrophobicity characteristics), and ligand binding sites. The altered cleaved site pattern and ubiquitination tendency support the hypothesis that the relatively lower expression of the GBE1 R198T mutant was due to degradation of misfolded GBE1 R198T mediated by the internal proteasome UPS and immunoproteasome pathways. In contrast, GBE1 Y329S conformational change did not cause a major change in the ligand-binding site and ubiquitination tendency, which correlates with Y329S structural stability6 and a lesser degradation activity, together explaining the milder clinical manifestation exhibited by Y329S patients.
In addition to molecular modeling, we performed an in vitro protein expression study to confirm the pathogenicity of the R198T variant. While multiple comparative tests only showed that all mutations were significantly lower than WT, we qualitatively noticed that the Y329S band signal was denser than that of L224P and R198T. This finding supports the fact that Y329S cases tend to have non-progressive or asymptomatic clinical symptoms compared to L224P with lethal early onset GSD IV.5,6 Reflecting from R198T clinical manifestation, which manifests at an early age, as well as the qualitative similarity of the R198T signal band with L224P, we conclude that the R198T novel mutation is more similar to L224P in terms of pathogenicity and clinical progression. Again, a similar pattern was observed between R198T and L224P in molecular modeling and in vitro studies, further strengthening the pathogenicity evidence for R198T.
To our knowledge, this study is the first to report and establish the pathogenicity of GBE1 R198T. Coincidentally, this is also the first functional study conducted to confirm the pathogenicity of a functional rare disease in Indonesia. We emphasize in silico mutation analysis, protein modeling, and robust protein expression functional studies by comparing novel VUS R198T, normal (WT), and pathogenic controls (L224P and Y329S), instead of focusing on clinical case reports. Reflecting on this GBE1 R198T case, 5 years have passed since the first patient onset in mid-2019, which was similar to the time taken to diagnose RD worldwide (5.6–7.6 years).11–13 GSD IV patients, whose phenotypes largely intersect with other hepatobiliary and hematologic diseases, may benefit from early genetic sequencing. Genetic sequencing, together with other clinical workups, such as liver and bone marrow biopsies, is more beneficial for diagnosing GSD IV. Early diagnosis, before liver damage, increases patient prognoses by allowing opportunities to decelerate disease progression by diet modification or organ transplant in the early stages of disease, as there is no enzyme replacement therapy or gene therapy available for GSD IV.
Our study had several limitations. First, the patient died in early 2022 owing to a suspected end-stage liver disease complication, and the patient’s guardian did not agree to an autopsy. Second, since several patient workups were physically documented on paper, it was unavoidable that some of the supporting documents (e.g., biopsy pictures) were unobtainable.
In conclusion, our study showed that a novel missense GBE1 mutation, R198T, is pathogenic in a patient with GSD IV with confirmed family inheritance. The amino acid replacement located in the central catalytic core was predicted to be damaging or disease-causing, based on in silico mutation analysis, molecular modeling, and ligand-protein docking, which was confirmed by the lower expression of GBE on Western blotting. The evidence confirmed the pathogenic nature of R198T, which was classified as likely pathogenic according to the ACMG criteria. Our study provides an example of how integrative in silico mutation prediction analysis, molecular modeling, protein-ligand docking, and in vitro functional studies can synergistically confirm the pathomechanism of novel mutation-causing functional rare diseases.
This study was approved by the Ethics Committee of the Faculty of Medicine, University of Indonesia – Cipto Mangunkusumo Hospital on 12 December 2022 (KET-1395/UN2.F1/ETIK/PPM.00.02/2022). Written informed consent was obtained from the patient’s parents for participation in this study and for the use of biological samples, including cryopreserved samples, for the experiments described in this research.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)