Keywords
eNOS, Primer Design, Rattus norvegicus, qPCR, Erectile Dysfunction, Diabetes Mellitus
This article is included in the Cell & Molecular Biology gateway.
eNOS, Primer Design, Rattus norvegicus, qPCR, Erectile Dysfunction, Diabetes Mellitus
Nitric oxide (NO) is a ubiquitous signaling molecule essential for the maintenance of diverse physiological and pathological processes in nearly all living organisms (Locascio et al., 2023). Within the vascular system, NO is synthesized from L-arginine by the nitric oxide synthase (NOS) family, which comprises three distinct isoforms: neuronal (nNOS), endothelial (eNOS), and inducible (iNOS). NO acts as a critical regulator of systemic functions, ranging from arterial blood pressure maintenance to the complex mechanics of penile erection (Forstermann & Sessa, 2011). Furthermore, within the central nervous system, NO modulates energy homeostasis, memory, and sexual behavior by acting in the paraventricular nucleus and the medial preoptic area (Melis & Argiolas, 2021).
In sexual physiology, NO induces the relaxation of smooth muscle tissue within the corpus cavernosum, facilitating increased blood flow to the penis and triggering the erectile response (Davies, 2015). Consequently, aberrations in NO synthesis can lead to erectile dysfunction (ED). Patients with cardiovascular risk factors—such as hypertension and diabetes mellitus—frequently exhibit endothelial dysfunction, characterized by the inability of the endothelium to produce sufficient bioactive NO (Forstermann & Sessa, 2011). Specifically, eNOS expression is significantly downregulated in Type 2 diabetes (Bahadoran et al., 2023), correlating with a high incidence of erectile disorders (Defeudis et al., 2021).
Given the rising global prevalence of diabetes, there is an urgent need for effective pharmacotherapies. Developing these products requires rigorous efficacy testing using model organisms such as the Sprague-Dawley strain of Rattus norvegicus (Bryda, 2013). A critical component of these studies is the molecular analysis of gene expression using quantitative PCR (qPCR), which offers high sensitivity and efficiency (Bustin, 2002). However, the reliability of qPCR depends on primer design (Chen et al., 2023). Therefore, this study aims to design and validate PCR primers for the eNOS gene to provide a standardized reference for evaluating novel treatments for erectile dysfunction in diabetic models.
All experimental procedures were approved by the Health Research Ethics Committee, Faculty of Medicine, University of Lampung (No. 3468/UN26.18/PP.05.02.00/2024), and conducted in accordance with the Guide for the Care and Use of Laboratory Animals. Adult male Rattus norvegicus (Sprague-Dawley strain) were obtained from a certified breeder, Taufik Rat and Mice (No. 324/TRM/SK/VI/2024). This strain is widely used in cardiovascular and urological research because of its stable physiological characteristics (Bryda, 2013; Nalbantoglu et al., 2024). Animals were acclimatized for seven days at the INALAB DNA facility (5°22′8.33″ S, 105°14′42.388″ E) under a 12-hour light/dark cycle before tissue collection. Rats were housed in wire-covered cages with hardwood shavings bedding (0.5–1 cm thickness), which was replaced every three days to maintain hygiene and reduce microbial contamination. Six animals were housed per cage under controlled room temperature and humidity, with indirect natural daylight exposure. Standard pellet feed and water were provided, with both replaced daily. Prior to tissue harvesting, rats were anesthetized by intraperitoneal injection of ketamine (80 mg/kg body weight). Euthanasia was performed by cervical dislocation under deep anesthesia in accordance with the American Veterinary Medical Association Guidelines for Animal Euthanasia (2020). This procedure was performed while the animal was in an erect state or during courtship. Tissue collection was performed immediately after euthanasia to preserve nitric oxide synthase (NOS) mRNA levels in the corpus cavernosum (Park et al., 1999).
Specific oligonucleotide primers targeting the endothelial nitric oxide synthase (eNOS) transcript were designed using the NCBI Primer-BLAST suite. The Rattus norvegicus NOS reference sequence (RefSeq: NM_021838.2) was used as the alignment template. Primer design parameters were set to a GC content of 40–60% and a melting temperature (Tm) of 55–60 °C to ensure optimal hybridization efficiency (Chen et al., 2023). Three candidate primer pairs (N1, N2, and N3) were selected for experimental validation based on the absence of predicted secondary structures and 3′ self-complementarity. All primers were synthesized by Integrated DNA Technologies (Singapore).
Total RNA was isolated from corpus cavernosum tissue using the easy-BLUE™ Total RNA Extraction Kit (Cat. No. 17061, iNtRON, Seongnam-si, South Korea). To preserve transcript integrity, tissue samples were cryo-pulverized in liquid nitrogen using a sterile mortar and pestle. The powdered tissue was then homogenized in 1 mL of easy-BLUE™ reagent. Phase separation was performed by adding 200 μL of chloroform (Cat. No. 1.02445.2500, Merck, Darmstadt, Germany), followed by centrifugation according to the manufacturer’s instructions. RNA was precipitated using 400 μL of isopropanol (Cat. No. 1.00993.2500, Merck, Darmstadt, Germany), as described in established protocols (Stephenson, 2016).
The RNA pellet was washed with 1 mL of 75% ethanol (Cat. No. 1.07017.2511, Merck, Darmstadt, Germany), gently mixed by inversion, and centrifuged at 10,000 rpm for 5 minutes. The supernatant was discarded, and the pellet was air-dried by inverting the microtube on clean tissue. RNA was then rehydrated in 30 μL of nuclease-free water (Cat. No. IBS-BW007A, iNtRON, Seongnam-si, South Korea) and dissolved by incubation at 55 °C for 45 minutes, with gentle tapping every 15 minutes.
Quality Assessment: RNA concentration and purity (A260/A280 ratio) were quantified using a NanoPhotometer® P300 (Implen, München, Germany). Only samples with a purity ratio of 1.8–2.0 were progressed to downstream applications. To eliminate genomic DNA (gDNA) interference—a common cause of false-positive results in qPCR—samples were treated with 1 μL of RNase-Free DNase I (Cat. No. DNS050, Geneaid, Taipei, Taiwan) at 37 °C for 15 minutes (Die & Roman, 2012).
First-strand cDNA was synthesized using the HiSenScript™ RH(−) RT PreMix Kit (Cat. No. 25087, iNtRON, Seongnam-si, South Korea). The reaction was prepared by adding 2 μL of RNA extract, 18 μL of nuclease-free water (Cat. No. IBS-BW007A, iNtRON, Seongnam-si, South Korea), 0.25 μL of random hexamer primers, and 0.25 μL of oligo (dT) primers to the upper tube, followed by thorough mixing. This system employs a ribonuclease H-deficient reverse transcriptase, which minimizes RNA template degradation during long-range synthesis and enhances full-length cDNA yield (Bustin et al., 2009). The reaction mixture, containing both random hexamer and oligo (dT) primers, was incubated in a LineGene Mini S thermal cycler (Bioer, Hangzhou, China) at 42 °C for 15 minutes, followed by enzyme inactivation at 85 °C for 10 minutes.
To empirically determine the optimal annealing temperature (Ta), high-resolution melting (HRM) analysis was performed using RealMOD™ Green W2 2× qPCR mix (Cat. No. 25350, iNtRON, Seongnam-si, South Korea) in a LineGene Mini S thermal cycler (Bioer, Hangzhou, China). The PCR reaction mixture consisted of 5 μL of cDNA, 10 μL of RealMOD™ Green W2 2× qPCR mix, 5 μL of nuclease-free water (Cat. No. IBS-BW007A, iNtRON, Seongnam-si, South Korea), 0.25 μL of forward and reverse primers (10 μM each). This reaction composition was applied to each of the three primer pairs. HRM is a highly sensitive post-PCR technique that monitors the dissociation behavior of DNA duplexes, enabling the detection of nonspecific amplification products and primer dimers with greater precision than standard melting curve analysis (Tong & Giffard, 2012). A temperature gradient ranging from 50 to 61 °C was applied, with a high-resolution ramp rate of 0.2 °C/s.
Conventional PCR was performed using i-Taq 2× PCR Master Mix (Cat. No. 25027, iNtRON, Seongnam-si, South Korea) in a LineGene Mini S thermal cycler (Bioer, Hangzhou, China). Each reaction contained 5 μL of cDNA, 10 μL of i-Taq 2× PCR Master Mix, 5 μL of nuclease-free water (Cat. No. IBS-BW007A, iNtRON, Seongnam-si, South Korea), 0.25 μL of forward and reverse primers (10 μM each). This reaction composition was applied to each of the three primer pairs. The optimized thermal cycling conditions consisted of an initial denaturation at 95 °C for 5 minutes, followed by 35 cycles of denaturation at 95 °C for 1 minute, annealing at 57 °C for 15 seconds, and extension at 72 °C for 40 seconds.
PCR amplification products were verified by agarose gel electrophoresis using a Mupid-exU electrophoresis system (Mupid, Tokyo, Japan) with a 1.2% agarose gel. The gel was prepared by dissolving 0.48 g of SeaKem® LE Agarose (Cat. No. 50004, Lonza, Walkersville, USA) in 40 mL of 1× TAE buffer (Cat. No. IBS-BT002–3, iNtRON, Seongnam-si, South Korea), followed by heating until the solution became fully dissolved and transparent. After cooling for approximately 30–40 minutes, 2 μL of RedSafe™ nucleic acid staining solution (Cat. No. 21141, iNtRON, Seongnam-si, South Korea) was added under low-light conditions. The solution was then gently mixed, poured into a casting tray, and fitted with a comb to form wells. Once the gel had solidified, 7 μL of Sizer™-100 DNA Marker (Cat. No. 24073, iNtRON, Seongnam-si, South Korea; 100–1500 bp) was loaded into one well, followed by 5 μL of PCR products loaded into the remaining wells. Electrophoresis was conducted at 100 V for 25 minutes.
Verified PCR products were subjected to bidirectional Sanger Sequencing (PT. Inti Kemika Sejahtera, Jakarta, Indonesia). Sequence accuracy and genomic fidelity were assessed by aligning raw electropherograms to the reference genome using BioEdit and the NCBI BLAST database (Altschul et al., 1990).
Computational analysis using NCBI Primer-BLAST yielded three potential primer pairs (N1, N2, and N3) targeting the eNOS mRNA sequence of Rattus norvegicus (Accession: NM_021838.2). All candidates exhibited optimal thermodynamic parameters, with GC content ranging from 45.45% to 57.89% and a melting temperature (Tm) difference between the forward and reverse primers of less than 2 °C. Notably, the N3 pair demonstrated superior predicted specificity with a 3′ self-complementarity score of 0.00, suggesting a low probability of primer-dimer artifacts. The detailed physicochemical properties are summarized in Table 1.
Total RNA extracted from the rat corpus cavernosum showed high integrity and concentration. Spectrophotometric analysis revealed RNA concentrations of 2590 ng/μL (Sample 1) and 2938 ng/μL (Sample 2). The purity ratios were 1.995 and 1.953, respectively, falling within the ideal range of 1.8–2.0. These results confirmed that the extracts were free from significant protein and organic solvent contamination, providing a high-quality template for subsequent cDNA library construction.
HRM analysis (Figure 1) was conducted across a temperature gradient (50–61 °C) to determine the optimal annealing temperature (Ta) empirically. The HRM profiles demonstrated successful amplification for all three primer pairs. The derivative melting curves showed stable amplicon formation with distinct peaks, indicating that the Ta range of 56.75–58.7 °C is highly conducive to primer-template hybridization.
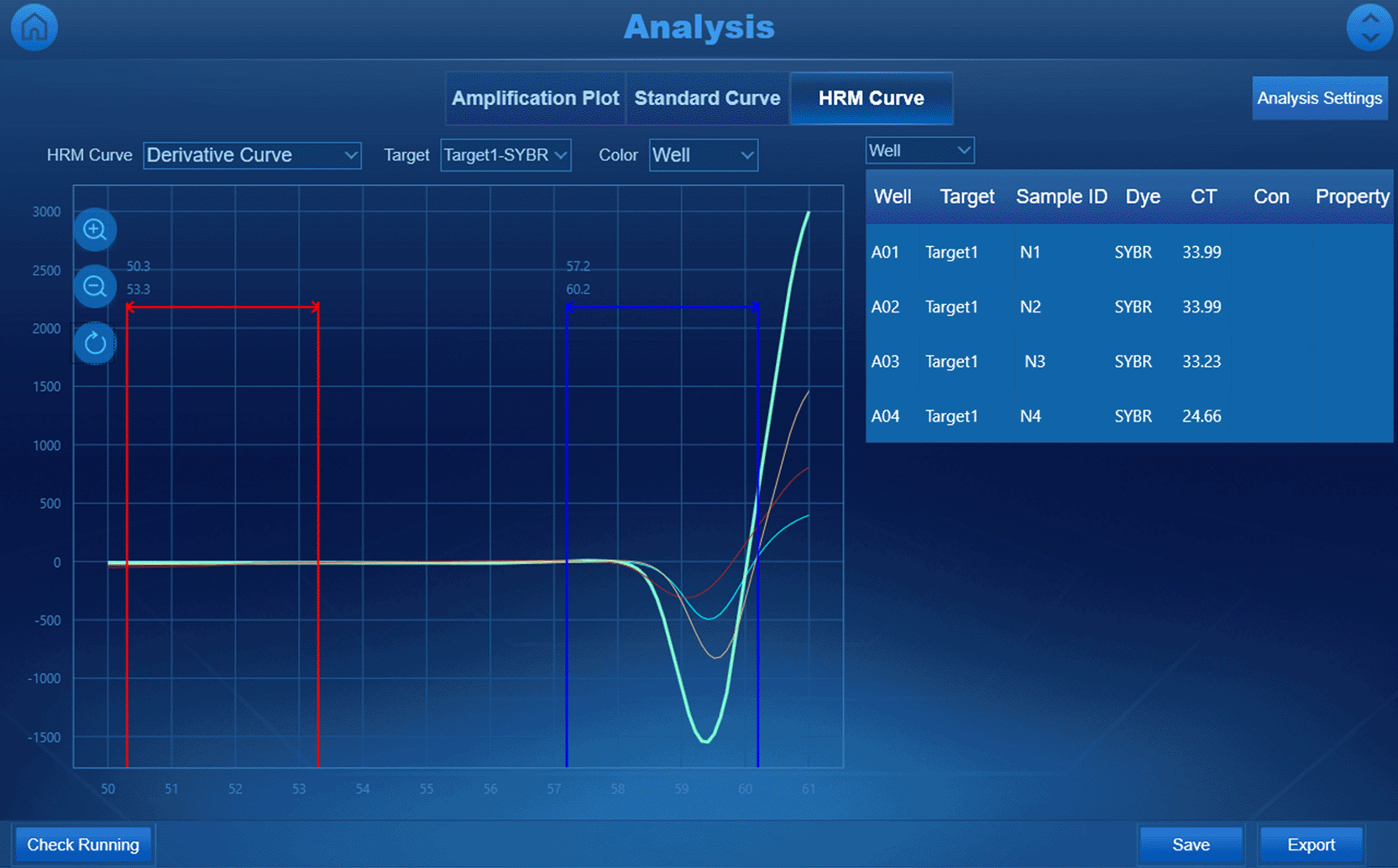
The derived HRM curves showed that fluorescence was most frequently detected between 56.75 °C and 58.7 °C, indicating that the optimal annealing temperature for primers N1, N2, and N3 is within this range. The presence of a single melting curve suggests that the primers specifically amplify the target sequence. Based on Ct values, although the differences were not significant, primer N3 showed slightly better amplification efficiency due to its lower Ct value.
The specificity of the amplicons was verified through 1.2% agarose gel electrophoresis. At a Ta 57 °C, all primer pairs produced single, high-intensity bands at the predicted molecular weights: N1 (869 bp), N2 (944 bp), and N3 (874 bp) (Figure 2). The absence of nonspecific bands or primer dimers at this temperature confirmed high amplification stringency.
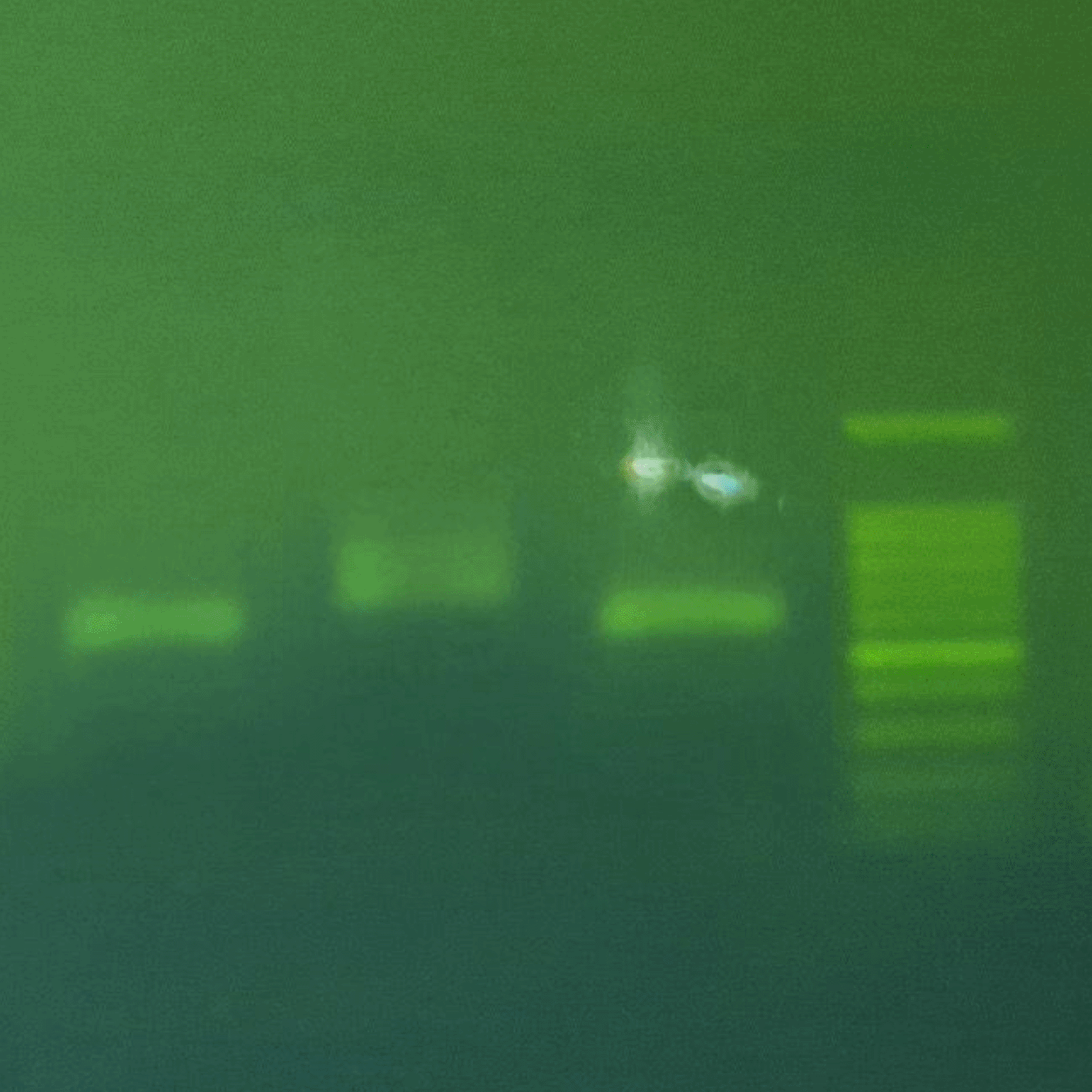
Description: N1: PCR results with primer pair N1 (±850 bp). N2: PCR results with primer pair N2 (±950 bp). N3: PCR results with primer pair N3 (±850 bp). MS: Marker Size (100–1000 bp).
In contrast, amplification at 59 °C yielded suboptimal profiles, characterized by multiple non-specific bands and low-molecular-weight primer dimers (Figure 3). This indicates that exceeding the optimized Ta significantly compromises the fidelity of the eNOS amplification.
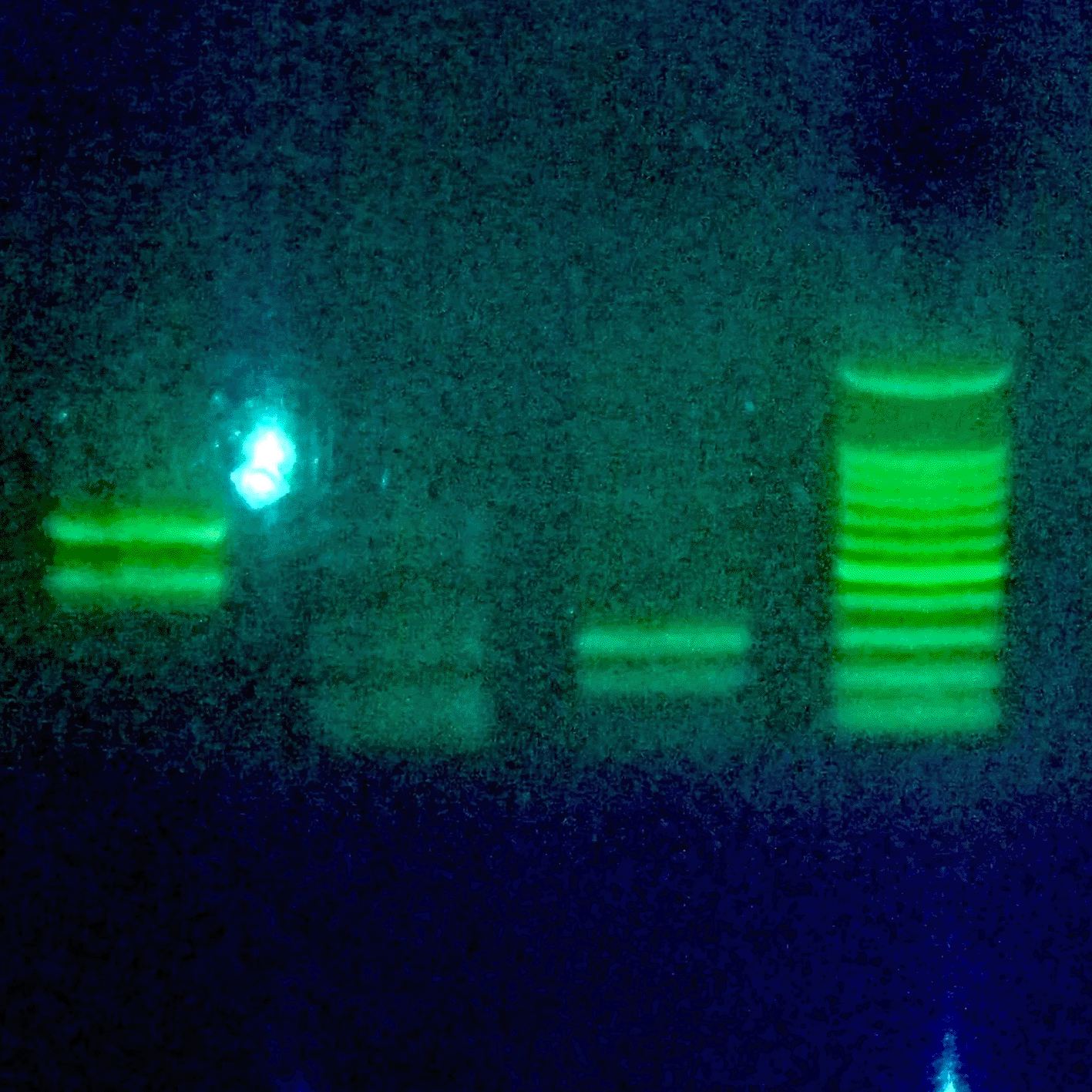
Description: N1: PCR results with primer pair N1 (±400 & 500 bp). N2: PCR results with primer pair N2 (±100 & 200 bp). N3: PCR results with primer pair N3 (± 200 & 300 bp). MS: Marker Size (100–1000 bp).
Validation through Sanger sequencing confirmed the identity of the PCR products (Figure 4). BLAST analysis revealed high homology between the amplicons and the Rattus norvegicus eNOS RefSeq (NM_021838.2). The N3 primer pair exhibited 100.00% identity, corresponding precisely to nucleotides 3104–3258 of the reference sequence ( Table 2). N1 and N2 showed 99.35% and 97.77% identity, respectively. Based on these findings, N3 was identified as the most reliable primer set for high-precision transcriptomic quantification.
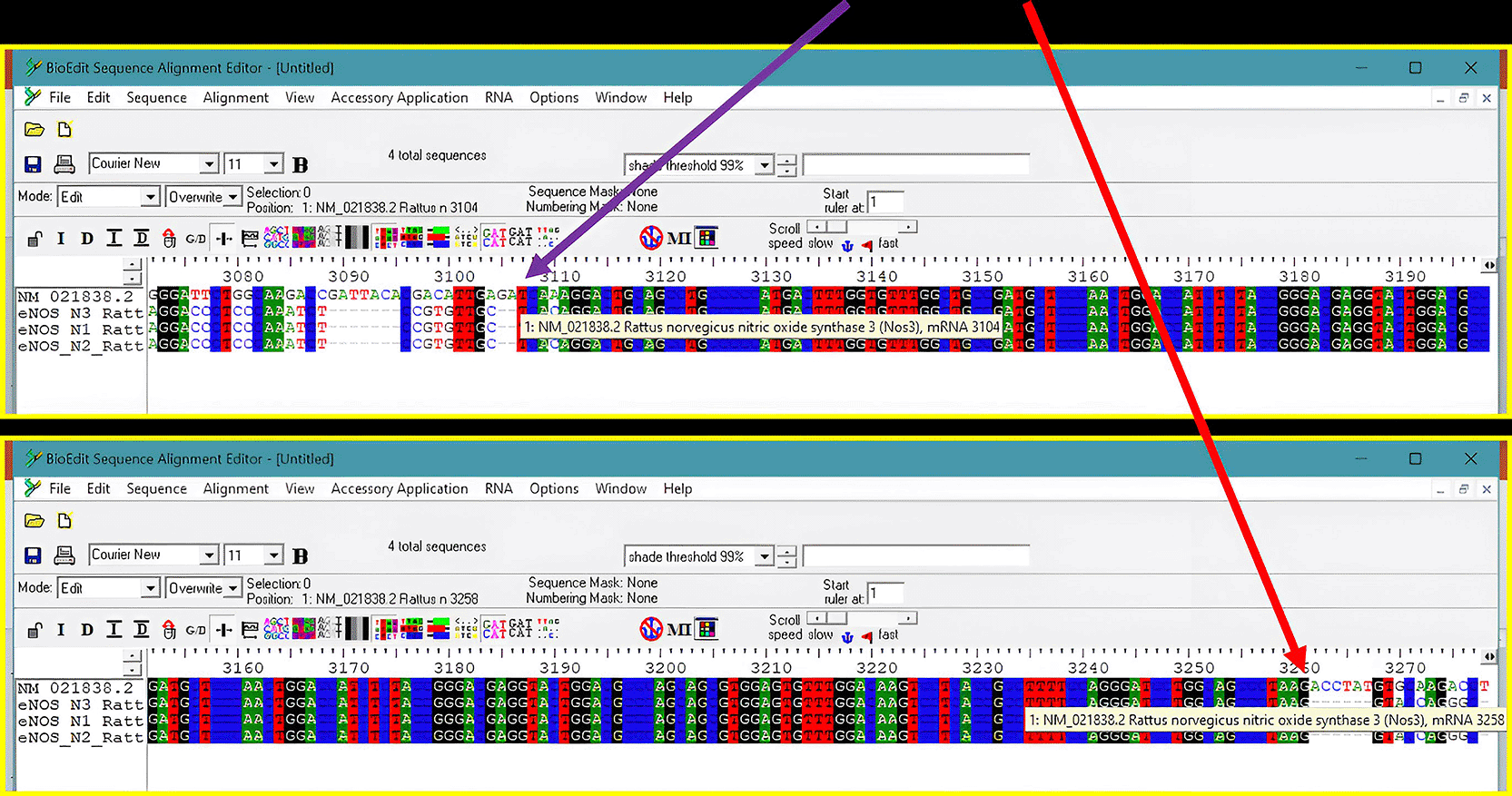
Sequencing results were used to verify primers N1, N2, and N3 through sequence alignment using BioEdit software with the reference sequence from NCBI (accession number NM_021838.2). Alignment of the four sequences showed that N1, N2, and N3 were conserved with the reference sequence at nucleotide positions 3104–3258.
Quantification of endothelial nitric oxide synthase (eNOS) mRNA is a critical diagnostic parameter for assessing vascular health, particularly in diabetic models, where the NO-cGMP pathway is significantly impaired. This study successfully validated a high-fidelity primer set, identifying the N3 pair as the optimal molecular tool for eNOS transcriptomic profiling.
The thermodynamic properties of the primer-template duplex fundamentally dictate the success of any qPCR-based assay. In this study, the N3 primer set exhibited an optimal GC content (52.63%) and a melting temperature (Tm) that enabled high-stringency binding. A critical observation was the absence of self-complementarity (Self 3′ value: 0.00). Theoretically, this minimizes the risk of primer-dimer formation, which can otherwise sequester reaction components and inflate fluorescence signals, leading to false-positive quantification (Chen et al., 2023).
By achieving a clean electrophoretic profile and a single-peak HRM dissociation curve, the N3 set ensures that the resulting Cq values reflect true biological variation rather than technical noise. This high level of specificity is essential because eNOS belongs to a family of related isoforms (nNOS and iNOS); thus, primers must be meticulously designed to avoid cross-amplification of non-target sequences (Forstermann & Sessa, 2011).
Experimental optimization identified 57 °C as the optimal annealing temperature (Ta). While computational tools like Primer-BLAST provide theoretical Ta values, the actual biochemical environment—including salt concentrations in the master mix and template complexity—requires empirical validation (Altschul et al., 1990).
The failure of amplification at 59 °C, characterized by non-specific “ghost bands” and dimers, illustrates the principle of “kinetic mismatching.” When the Ta approaches or exceeds the Tm of the primer-template duplex, the system’s kinetic energy may exceed the hydrogen bond strength of the specific target, preventing stable hybridization. Under these conditions, free primers are more likely to interact with each other or bind to secondary, lower-affinity sites on the cDNA template (Bustin et al., 2009). The successful amplification at 57 °C represents a precise balance between the entropy of primers in solution and the enthalpy of binding to the target eNOS sequence.
In diabetic models, hyperglycemia-induced oxidative stress leads to eNOS uncoupling, resulting in decreased mRNA stability and bioactive NO production (Bahadoran et al., 2023). Using a primer set with 100.00% identity (NM_021838.2) is essential to ensure that research on novel pharmacotherapies—such as those involving piperine or natural antioxidants—is based on accurate molecular data (Nalbantoglu et al., 2024).
The application of High-Resolution Melting (HRM) analysis provides an added layer of reliability. Unlike conventional melting curve analysis, HRM is sensitive enough to detect single-nucleotide polymorphisms (SNPs) and subtle variations in amplicon composition. This ensures that the N3 primer set remains robust across different Sprague-Dawley colonies, where minor genetic drift may occur (Tong & Giffard, 2012).
While mRNA quantification is a powerful tool for gene expression profiling, it is important to note that eNOS activity is also heavily regulated by post-translational modifications, such as phosphorylation at Ser1177. Therefore, while the N3 primer set offers a validated framework for transcriptomic analysis, future studies should complement these findings with Western Blotting for protein quantification and Griess assays for measuring total nitrate/nitrite levels to provide a holistic view of endothelial function (Defeudis et al., 2021).
In conclusion, this study successfully designed, optimized, and validated three primer pairs for the eNOS gene in Rattus norvegicus (Sprague-Dawley strain). Based on molecular analysis, an annealing temperature (Ta) of 57 °C was identified as optimal for specific amplification, with high thermal stability and no detectable primer-dimer formation.
Among the candidates, the N3 primer pair (F: 5’-GGCAAGACCGATTACACGA-3′; R: 5’-CAGTCCATCAAAGCATACGAAG-3′) demonstrated the highest specificity, yielding a 100.00% identity match with the GenBank reference sequence (NM_021838.2). The N1 and N2 primers also showed high reliability with 99.35% and 97.77% identity, respectively.
Given its perfect sequence alignment and clean electrophoretic profile, the N3 primer is highly recommended as a standardized molecular tool for RT-qPCR assays. This validated primer set provides a reliable foundation for future research investigating eNOS gene expression pathways and evaluating the efficacy of pharmacotherapies in treating diabetes-induced erectile dysfunction.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)