Keywords
Climate change, sustainability, functional genomics, beneficial microbes, Arabian Desert
This article is included in the Genomics and Genetics gateway.
This article is included in the Climate gateway.
Climate change, sustainability, functional genomics, beneficial microbes, Arabian Desert
Microorganisms inhabiting extreme environments, or extremophiles, have long served as models for studying the boundaries of life. They flourish in habitats once considered inhospitable, including icy polar regions, volcanic vents, acidic lakes, and arid deserts—through mechanisms such as specialized membrane lipids, molecular chaperones, metal resistance systems, and the production of protective pigments and exopolysaccharides (Neilson et al., 2012; Feller, 2017; Vieille & Zeikus, 2001). For example, psychrophilic species synthesize antifreeze proteins and cold-active enzymes to maintain metabolic activity under freezing conditions. At the same time, thermophiles adapt to high temperatures through thermostable proteins and ether-linked membrane lipids (Kates, 1992). Acidophiles such as Acidithiobacillus ferrooxidans resist toxic metal concentrations through efflux systems and biofilm formation (Dopson & Holmes, 2014). Likewise, desert-adapted microorganisms synthesize carotenoids and melanins that protect against UV radiation and produce extracellular polysaccharides (EPS) to retain moisture and mitigate desiccation stress (Cockell & Knowland, 1999; Sher et al., 2019). These mechanisms reflect evolutionary strategies that sustain microbial survival and support their host plant in the most arid habitats.
In arid ecosystems, plants depend on their associated microbiota to tolerate abiotic stress and maintain productivity. The Arabian Peninsula is among the driest regions globally, hosting a limited number of highly adapted plant species that thrive under extreme temperature, salinity, and water scarcity. These plants exhibit physiological traits such as deep root systems, osmoprotectant accumulation, and efficient water-use strategies that allow persistence in nutrient-poor soils. However, their resilience is also shaped by intimate interactions with their endophytic microbiota, defined as microorganisms residing inside plant tissues without causing harm (Alsharif et al., 2020). Endophytic bacteria enhance plant growth by producing phytohormones, solubilizing nutrients, and synthesizing bioactive metabolites that improve drought and salinity tolerance (Fanai et al., 2024) They also contribute to nitrogen fixation and carbon cycling, processes vital for sustaining life in desert ecosystems where organic matter is limited (Feiner et al., 2015).
Despite their ecological and potential biotechnological importance, the desert plant microbiome, particularly endophytic communities, remains poorly characterized. Previous studies in other desert systems, such as the Atacama and Namib, revealed microbial lineages uniquely adapted to aridity and nutrient limitation (Ronca et al., 2015) but similar research in the Arabian Peninsula is limited. Existing data on desert soils and rhizospheres provide limited insight into the microbiota living within plant tissues, which are likely to harbor distinct communities with specialized functional roles (Turner et al., 2013). Understanding the structure and function of these plant-associated microbial communities is therefore critical to elucidating how desert plants persist under extreme abiotic stress and to identify microbial traits that can be leveraged to enhance agricultural resilience in drylands.
Recent advances in culture-independent methods, particularly high-throughput 16S rRNA gene sequencing and metagenomic inference tools such as PICRUSt2, now enable comprehensive profiling of microbiomes across ecosystems (Haryono et al., 2021). These tools allow simultaneous assessment of microbial diversity and prediction of metabolic pathways involved in nutrient metabolism, stress tolerance, and environmental detoxification. Applying such approaches to native desert plants can uncover microbial taxa and genes that facilitate survival in environments projected to expand under global warming. Moreover, desert-derived microbes have emerged as promising sources of enzymes, pigments, and osmoprotectants that could be harnessed for biotechnology and sustainable agriculture (Ortiz et al., 2021).
In this study, we investigated the endophytic bacterial communities associated with four desert plants native to the Arabian Peninsula namely Zygophyllum mandavillei, Tribulus zeyheri, Limeum arabicum, and Cyperus conglomeratus. Z. mandavillei is native to hyper-arid zones of the Arabian Peninsula and represents a model for studying drought tolerance and symbiotic adaptation (Abdullah, 2017) T. zeyheri (Zygophyllaceae) is known to promote sand fixation and water conservation (Khan et al., 2020). L. arabicum, native to the Arabian deserts, is known for its medicinal properties and tolerance to extreme aridity (Khan et al., 2020). C. conglomeratus (Cyperaceae), abundant across the Middle East and North Africa, is valued for its anti-inflammatory properties and capacity to thrive in sandy and nutrient-poor soils (Brown & Feulner, 2023). These plant systems collectively provide an ideal lens to study microbial resilience under increasing global aridification. Using 16S rRNA gene amplicon sequencing and functional inference analysis, we aimed to (1) characterize their microbial diversity and taxonomic composition, (2) identify dominant and unique taxa associated with each host plant, and (3) predict the metabolic pathways that enable microbial survival under extreme desert conditions. By linking microbial diversity with functional potential, this study reveals ecological strategies that support desert ecosystem resilience and demonstrates how native plant microbiomes can guide the development of microbial solutions for sustainability and climate adaptation.
Four desert plant species native to the Arabian Peninsula namely Zygophyllum mandavillei, Tribulus zeyheri Sond., Limeum arabicum, and Cyperus conglomeratus Rottb. were selected for microbiome analysis, as these species are highly adapted to arid environments and play key roles in desert ecosystem stability. The plant material used in this study was identified by Dr Walaa Mousa. A voucher specimen was not deposited for this study because no new collection was made; the species is already maintained and documented at the UAE Plant Genetic Resource Centre, and live specimens of the same species are also demonstrated in the educational botanical garden in Al Ain City.
During November 2024, plant material was collected from Al-Ain desert, United Arab Emirates (approximate coordinates: 24.20° N, 55.70° E). At each of five independent adjacent sites per plant species, 6-7 individual plants were sampled and then pooled to form one composite sample. Thus, for the four species across five sites, a total of 130 plant samples (4 species × 5 sites × 6-7 independent plant samples, each sample is a composite of 5 pooled subsamples) were obtained. Samples were placed in sterile polyethylene bags, transported on ice, and transitely stored at −20°C until further processing.
Endophytic (endosphere) microbiota were analyzed following modifications of protocols by Coombs & Franco (Coombs & Franco, 2003) and Edwards et al. (Edwards et al., 1991). Plant tissue were surface-sterilized by immersion in 95% ethanol for 3 min, rinsed with sterile distilled water, immersed in 3% sodium hypochlorite for 5 min, followed by three sterile water rinses. Sterilization was repeated twice to ensure efficacy. To validate surface sterilization, 100 μL of the final rinse water was plated on tryptic soy agar (TSA) and incubated at 25°C and 37°C for 72 h; absence of colony growth confirmed sterilization. Sterile tissues were ground under liquid nitrogen, and total genomic DNA was extracted using the chloroform–isoamyl alcohol method (Edwards et al., 1991). DNA quality was verified by 1% agarose gel electrophoresis and quantified using a NanoDrop™ spectrophotometer (Thermo Fisher Scientific, USA).
The V3–V4 hypervariable region of the bacterial 16S rRNA gene was amplified using primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 785R (5′-GACTACHVGGGTATCTAATCC-3′) (Klindworth et al., 2013). These primers provide broad bacterial coverage (~96%) with minimal bias (Thijs et al., 2017) (Fadeev et al., 2021). Each 50 μL PCR reaction comprised ~30 ng template DNA, 1× Taq buffer, 4 mM MgCl2, 0.2 mM dNTPs, 0.5 μM each primer, and 1 U Taq DNA polymerase. Cycling conditions: initial denaturation at 94°C for 3 min; 30 cycles of 94°C for 30 s, 56°C for 45 s, 72°C for 45 s; final extension at 72°C for 10 min. Amplicons were visualized on 1.5% agarose gel, purified using QIAquick PCR & Gel Cleanup Kit (QIAGEN, Germany), quantified with the PicoGreen dsDNA assay (Thermo Fisher Scientific), and then pooled equimolarly. Clean-up of pooled amplicons was conducted using AMPure XP beads (Beckman Coulter, USA).
Library preparation followed standard Illumina MiSeq protocols (Caporaso et al., 2012). Library size distributions were checked on an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Paired-end sequencing (2 × 250 bp) was performed at BGI (Shenzhen, China). Raw FASTQ data have been deposited in the NCBI Sequence Read Archive upon manuscript acceptance (accession to be added).
Demultiplexed paired-end FASTQ reads were processed using DADA2 (v1.30.0) in R (v4.3.2) (Callahan et al., 2016). Forward and reverse reads were trimmed to remove adapters and low-quality bases; error rates were learned using learnErrors(), ASVs inferred with dada(), merged via mergePairs(), and chimeras removed with removeBimeraDenovo(). Taxonomic assignment was carried out against the Genome Taxonomy Database (GTDB release R207) (Parks et al., 2022). The resulting ASV tables were used for downstream ecological and statistical analysis.
Functional potential of ASVs was inferred using PICRUSt2 (v2.3.0-b) (Douglas et al., 2020). Predicted gene families were mapped to KEGG Orthology (KO), Clusters of Orthologous Groups (COG), and MetaCyc pathways. Differential enrichment among plant hosts was assessed using Wilcoxon rank-sum and Kruskal–Wallis tests.
To account for sequencing depth variation, ASV tables were rarefied using the vegan package (v2.7-2) (Oksanen et al., 2025) in R v4.3.2. Alpha-diversity metrics (Observed, Chao1, ACE, Shannon, Simpson, Good’s coverage) were calculated using phyloseq (v1.46.0) (McMurdie & Holmes, 2013) and visualized with ggstatsplot (v0.12.1) (Patil, 2021). Beta-diversity analyses used Bray–Curtis, unweighted and weighted UniFrac distances; ordinations (nMDS and PCoA) were generated via QIIME v1.8.0 (Caporaso et al., 2012). For PCoA, 100 iterations of random subsampling (75% of the smallest library size) were used to ensure stability of ordination plots. Group differences were tested using PERMANOVA (p < 0.05). Dominant taxa at phylum and class levels were visualized using plot_bar in phyloseq. All statistical analyses were conducted in R v4.3.2 and figures were produced using ggplot2 (Wickham, 2016).
This study examined the endophytic bacterial communities of four desert plant species native to the Arabian Peninsula, namely Z. mandavillei (A), T. zeyheri (B), L. arabicum (C), and C. conglomeratus (D) ( Figure 1). These species are dominant components of the desert flora in the United Arab Emirates and exhibit exceptional tolerance to heat, salinity, and drought.

(A) Zygophyllum mandavillei, (B) Tribulus zeyheri Sond., (C) Limeum arabicum, and (D) Cyperus conglomeratus Rottb. These four native plants of the Arabian Peninsula were selected for microbiome profiling to explore their endophytic bacterial diversity and functional potential under arid conditions.
A total of 110 root samples were analyzed, representing four plant species collected from five independent desert sites, with each site sample derived from a composite of 5 subsamples. Using 16S rRNA gene amplicon sequencing, we characterized the taxonomic composition, diversity, and predicted functional potential of their endospheric microbiomes. After quality control and removal of host-derived reads, a total of 3,660,664 high-quality sequences were obtained. Clustering at 97% similarity revealed 30 bacterial phyla, 15 classes, 20 orders, 27 families, and 21 genera. Rarefaction analysis indicated that sequencing depth was sufficient to capture the microbial diversity within each plant species ( Figure 2). The curves reached a plateau, confirming that read coverage adequately represented the bacterial communities associated with these samples.
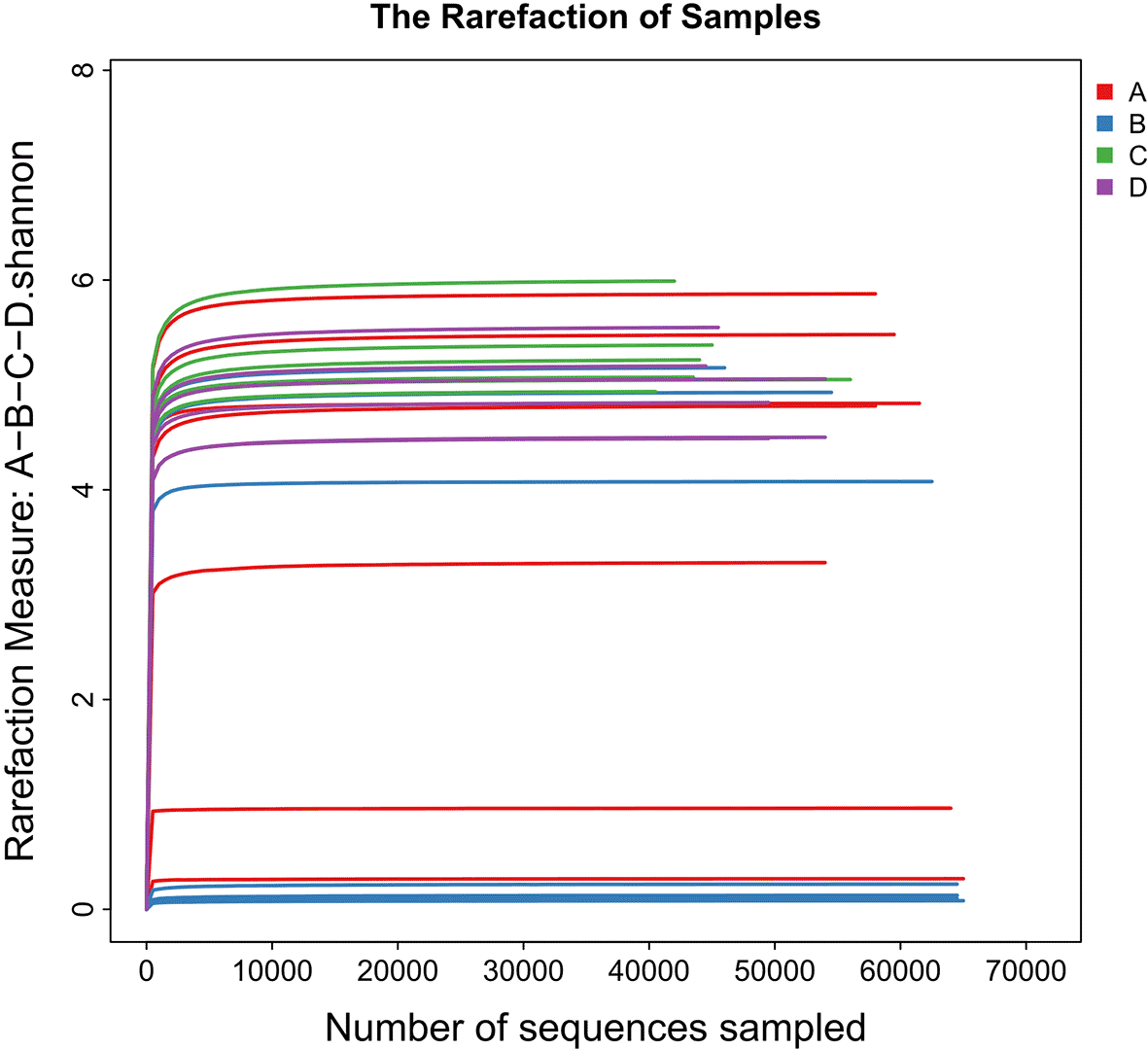
Curves represent the relationship between sequencing depth and the number of observed amplicon sequence variants (ASVs) for each plant species.
To assess richness among the four desert plant species, alpha diversity was evaluated using six indices: Observed species, Chao1, ACE, Shannon, Simpson, and Good’s coverage ( Figure 3). All richness-related indices (Observed, Chao1, and ACE) showed significant differences among species (p < 0.05), with Z. mandavillei (A) exhibiting the highest richness, followed by L. arabicum (C) and C. conglomeratus (D), while T. zeyheri (B) displayed the lowest values. This indicates that Z. mandavillei supports a more diverse and taxonomically rich bacterial community than T. zeyheri.
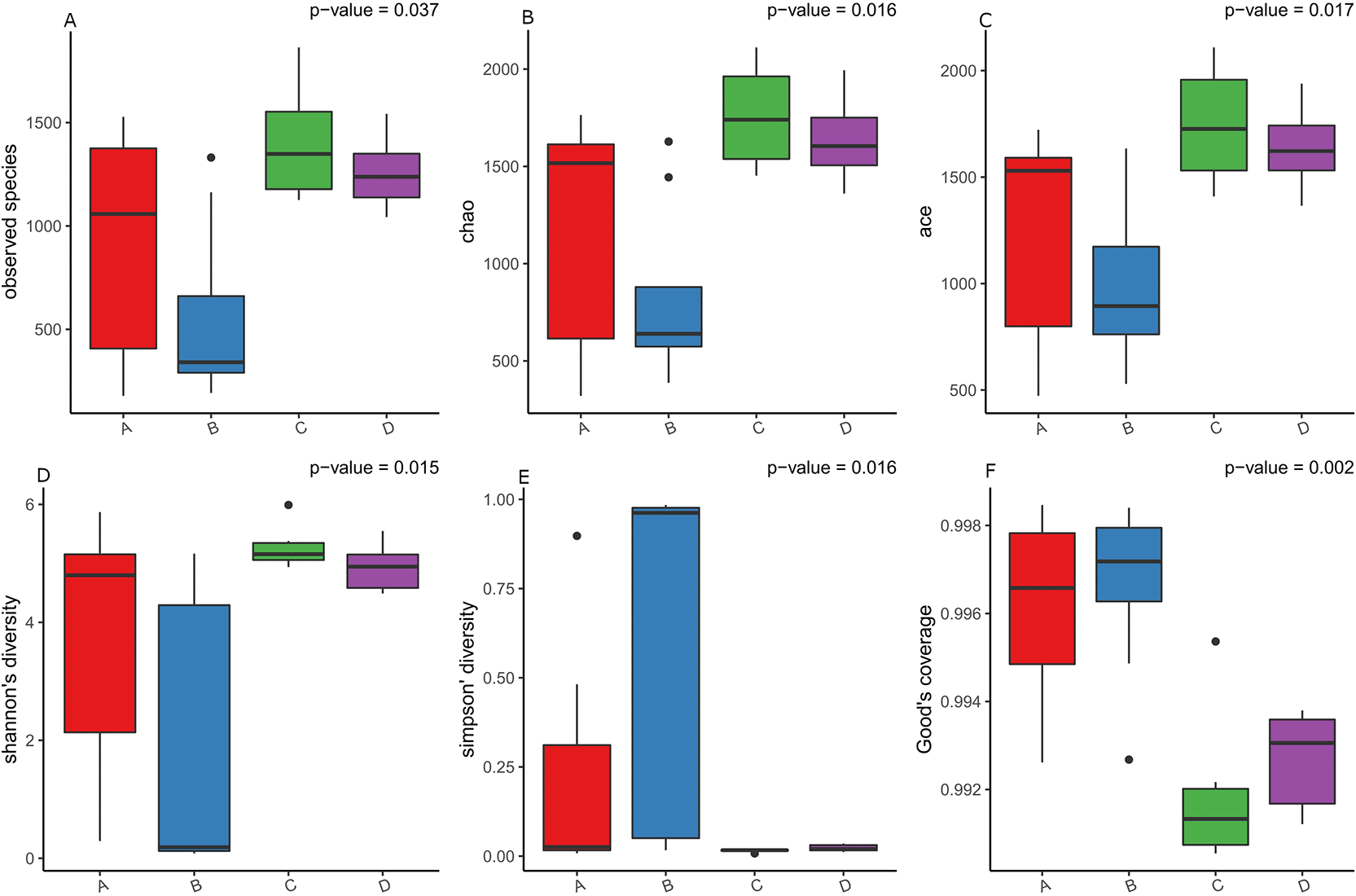
Six diversity indices were applied to assess species richness and evenness: (3-A) Observed index (number of detected species), (3-B) Chao1 (estimated species richness), (3-C) ACE (community composition variability), (3-D) Shannon index (species richness and evenness), (3-E) Simpson index (dominance and evenness), and (3-F) Good’s coverage (sampling completeness).
Diversity and evenness metrics (Shannon and Simpson) revealed distinct community structures across hosts. Shannon diversity was higher in Z. mandavillei (A), reflecting balanced taxa distribution, whereas T. zeyheri (B) showed low Shannon but high Simpson values, suggesting dominance by a few abundant taxa. Overall, each plant maintained a distinct endophytic bacterial diversity profile, with Z. mandavillei (A) harboring the richest and most even community, and T. zeyheri (B) showing reduced diversity, possibly due to host-specific selection or differing root microenvironments.
Beta diversity analysis was performed to compare the overall microbial community composition among the four desert plant species. Three distance metrics were applied, including Bray–Curtis dissimilarity, weighted UniFrac, and unweighted UniFrac in order to capture both taxonomic and phylogenetic differences ( Figure 4). The Bray–Curtis dissimilarity index revealed a highly significant difference among the four groups (p = 0.001), indicating that each plant species harbors a distinct endophytic bacterial composition. This separation reflects strong host-specific effects on community assembly rather than random variation. Unweighted UniFrac analysis, which accounts for the presence or absence of taxa, supported these findings and emphasized that community composition differs substantially between hosts. The weighted UniFrac metric, which incorporates relative abundances, further indicated that compositional variation arises from both dominant and rare taxa, suggesting host-driven selection shaping the bacterial communities and demonstrating a pronounced dissimilarity in microbiome structure among the four desert plant species.
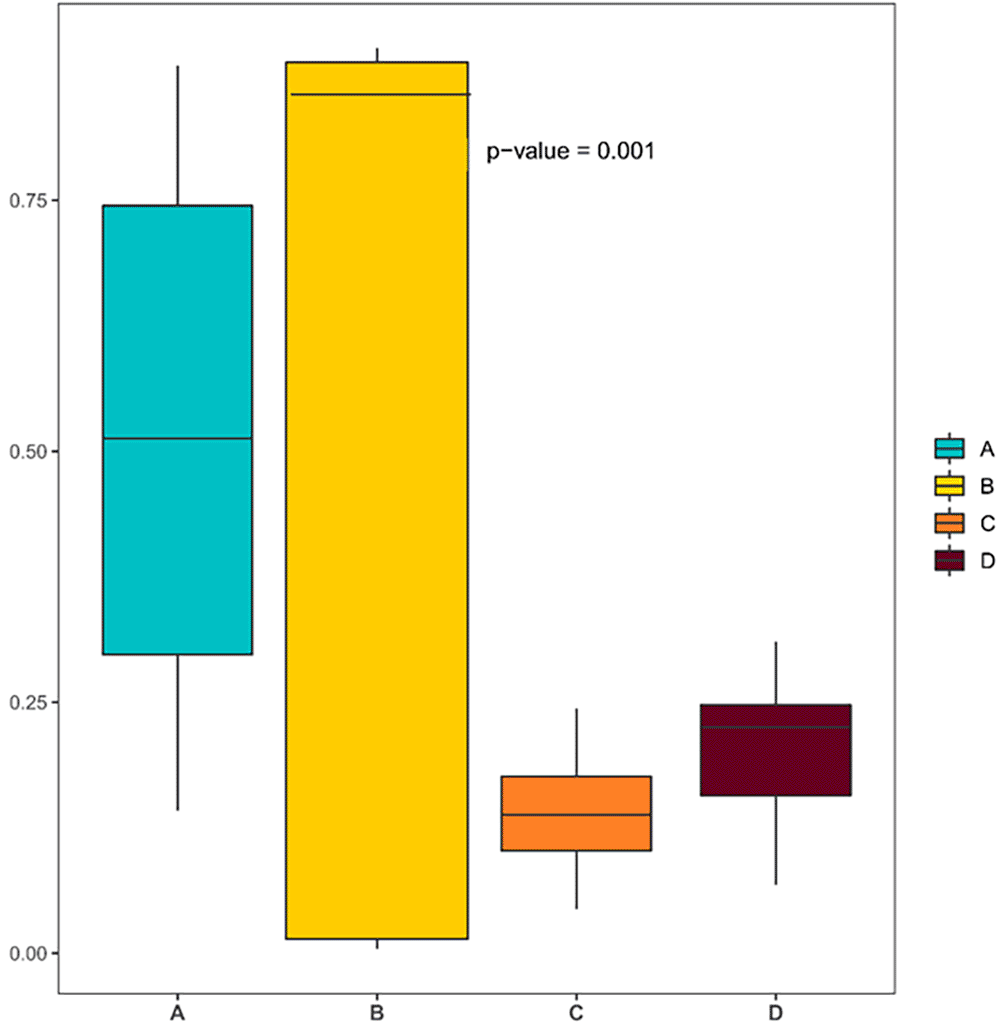
The Bray–Curtis dissimilarity metric was used to assess compositional differences between samples, revealing distinct clustering patterns and high dissimilarity among the microbial communities associated with each plant species.
Principal Coordinates Analysis (PCoA) based on unweighted UniFrac distances was used to visualize compositional differences in endophytic bacterial communities among the four desert plant species ( Figure 5). The first two principal coordinates explained 36.51% and 11.28% of the total variance, respectively. The ordination plot revealed clear clustering by plant host (adonis R² = 0.30, p = 4 × 10−4), confirming that community composition differs significantly among species. Z. mandavillei (A) and T. zeyheri (B) formed distinct, non-overlapping clusters, indicating highly specialized bacterial assemblages. In contrast, the microbiomes of L. arabicum (C) and C. conglomeratus (D) clustered closely together, suggesting they share a larger proportion of bacterial taxa and potentially similar ecological or physiological drivers of colonization. Taken together, these results point to a strong host effect in structuring the desert plant microbiome. While each species supports a characteristic bacterial consortium, the partial overlap between L. arabicum and C. conglomeratus implies that some microbial lineages may thrive across hosts with comparable root habitats or resource profiles.
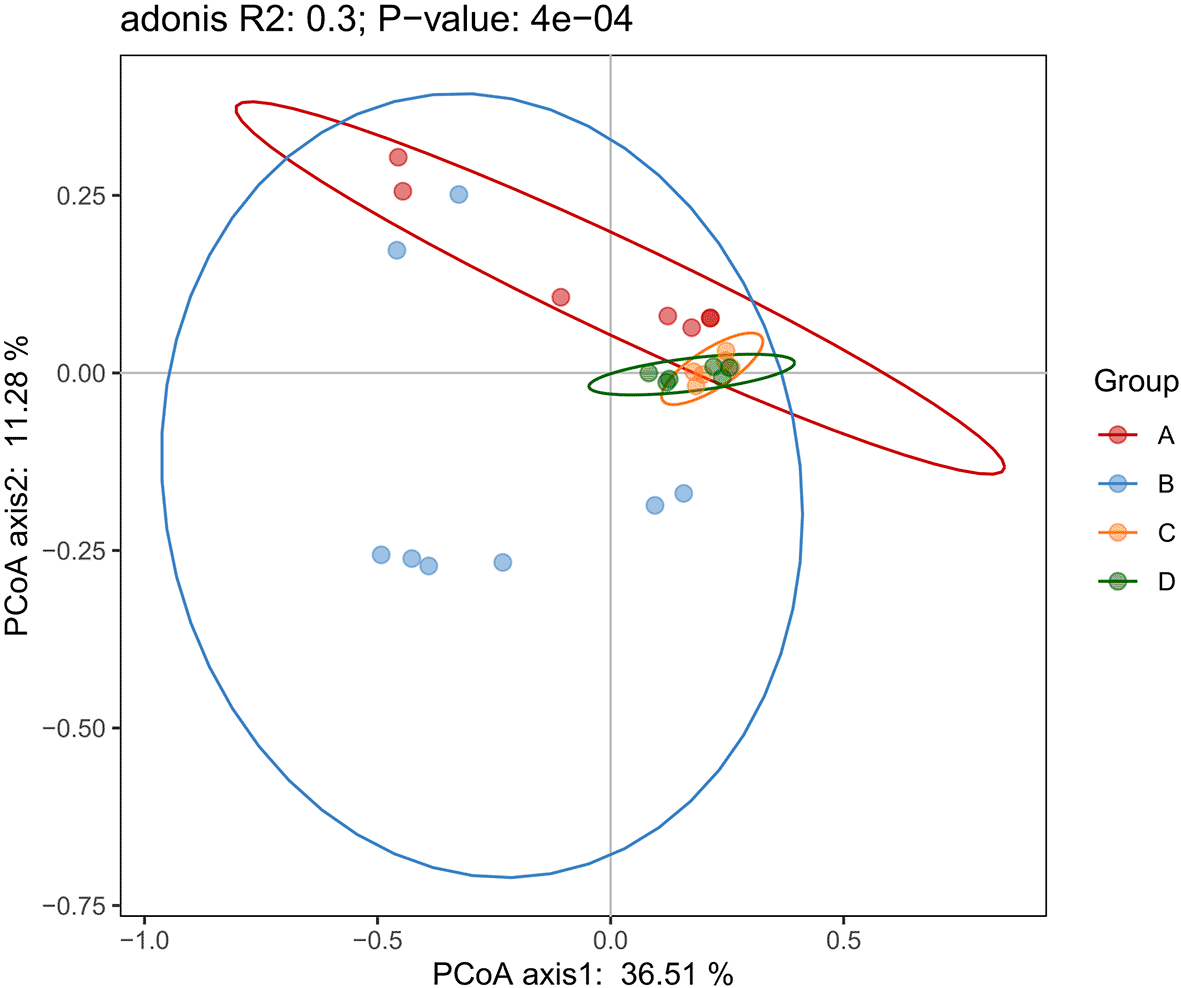
The analysis depicts phylogenetic beta diversity among microbial communities associated with the four desert plant species. Distinct clustering patterns indicate clear differences in community composition, reflecting variations in microbial structure and phylogenetic relationships across samples.
To evaluate phylogenetic differences in the bacterial communities among the four desert plant species, a weighted UniFrac-based Principal Coordinates Analysis (PCoA) was conducted ( Figure 6). The first two axes explained 82.6% and 5.65% of the total variance, respectively, accounting for 88.25% of the variation in the dataset.
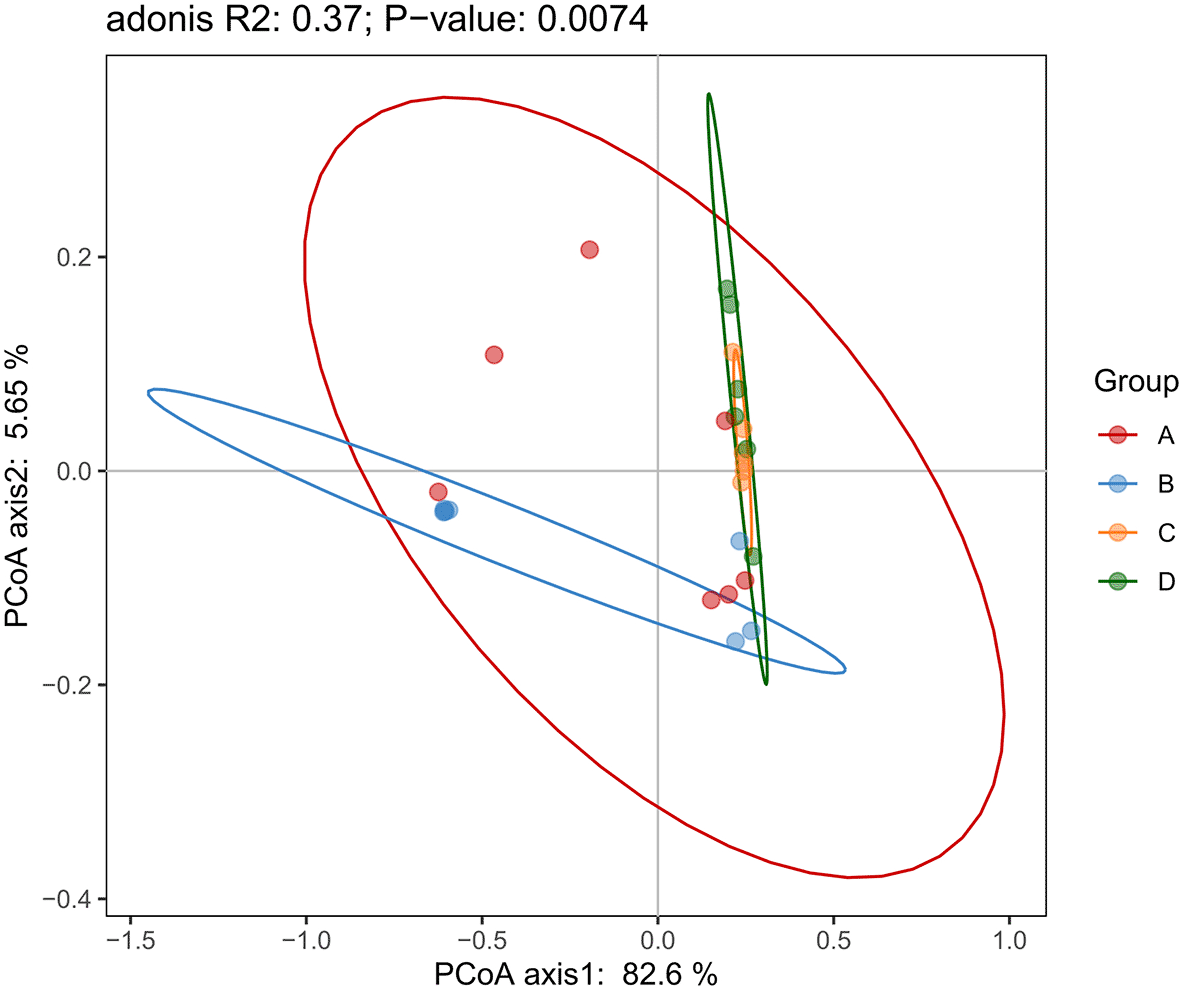
This analysis visualizes beta diversity among the microbial communities of the four desert plant species, incorporating both phylogenetic relationships and relative abundance of taxa. Distinct clustering patterns reflect compositional and abundance-based differences in microbial communities across samples.
The ordination revealed a statistically significant separation among the plant-associated bacterial communities (adonis R² = 0.37; p = 0.0074), indicating that host identity strongly influences microbiome composition. Group A formed a well-defined cluster along PCoA axis 1, distinct from all other groups. Group B was positioned separately on the same axis, showing partial proximity to A but maintaining a unique composition. Groups C and D overlapped substantially, suggesting similarity in their endophytic bacterial communities; however, the slight displacement of D indicates minor but significant differences in community structure. These results demonstrate that host plant species play a major role in shaping bacterial community composition and phylogenetic structure, with Z. mandavillei (A) and T. zeyheri (B) harboring particularly distinct microbiomes, while L. arabicum and C. conglomeratus (C and D) share a more comparable microbial profile.
The microbial communities associated with the studied plant species displayed distinct taxonomic profiles across multiple taxonomic levels ( Figure 7). At the phylum level, all groups were dominated by Pseudomonadota, followed by Cyanobacteriota, Actinomycetota, and Gemmatimonadota. Pseudomonadota was consistently abundant in all plants, while Cyanobacteriota was particularly enriched in Z. mandavillei (A) and T. zeyheri (B). In contrast, Actinomycetota and Gemmatimonadota were more prevalent in L. arabicum (C) and C. conglomeratus (D), suggesting adaptation of these taxa to more nutrient-limited microhabitats. At the class level, Alphaproteobacteria dominated all four plants, confirming its central role as an endophytic lineage in desert ecosystems. Cyanophyceae was highly abundant in Z. mandavillei (A) and T. zeyheri (B) but nearly absent in L. arabicum (C) and C. conglomeratus (D). Gammaproteobacteria was enriched in Z. mandavillei (A), whereas Actinobacteria and Betaproteobacteria were markedly more abundant in L. arabicum (C) and C. conglomeratus (D), reflecting host-specific colonization preferences.
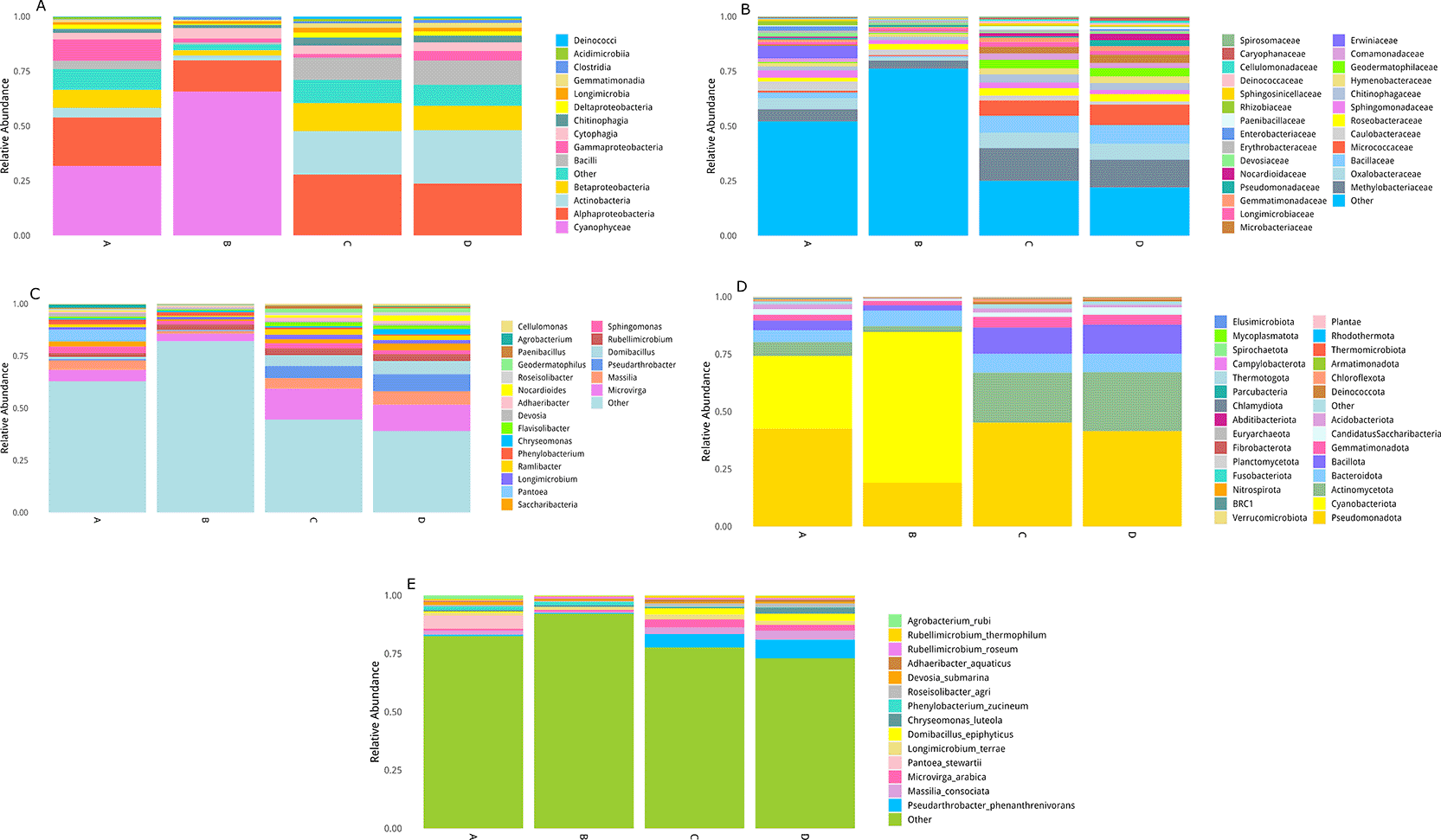
The relative abundance of bacterial taxa is shown at multiple taxonomic levels: (A) class, (B) genus, (C) family, (D) phylum, and (E) species. The data illustrate distinct microbial community structures among the analyzed plants, highlighting dominant taxa contributing to functional and ecological diversity within the desert endosphere.
At the family level, Methylobacteriaceae was among the most abundant families across all plant hosts, consistent with its known association with plant tissues. Z. mandavillei (A) exhibited additional enrichment of Oxalobacteraceae, Bacillaceae, Sphingomonadaceae, Erwiniaceae, and Caulobacteraceae. T. zeyheri (B) was dominated by Methylobacteriaceae, Roseobacteraceae, and Sphingomonadaceae. L. arabicum (C) and C. conglomeratus (D) showed high relative abundance of Methylobacteriaceae, Oxalobacteraceae, Bacillaceae, and Micrococcaceae, families commonly associated with stress tolerance and nutrient cycling.
At the genus level, Microvirga, Massilia, and Domibacillus were enriched in Z. mandavillei (A), while Pseudarthrobacter dominated the communities of L. arabicum (C) and C. conglomeratus (D). T. zeyheri (B) exhibited lower overall diversity, with Methylobacterium and Sphingomonas as the main representatives. At the species level, Pseudarthrobacter phenanthrenivorans, Massilia consociata, Microvirga arabica, and Domibacillus epiphyticus were predominant in L. arabicum (C) and C. conglomeratus (D), indicating high similarity between their microbial communities. In contrast, Z. mandavillei (A) showed unique enrichment of Pantoea stewartii and Agrobacterium rubi, suggesting distinct host–microbe interactions and possibly specialized metabolic associations.
To assess the potential metabolic activity of the analyzed microbiota, predicted functional profiling of the endophytic bacterial communities was performed using PICRUSt2, and annotated using the MetaCyc, Clusters of Orthologous Groups (COG), and KEGG Orthology (KO) databases ( Figure 8).
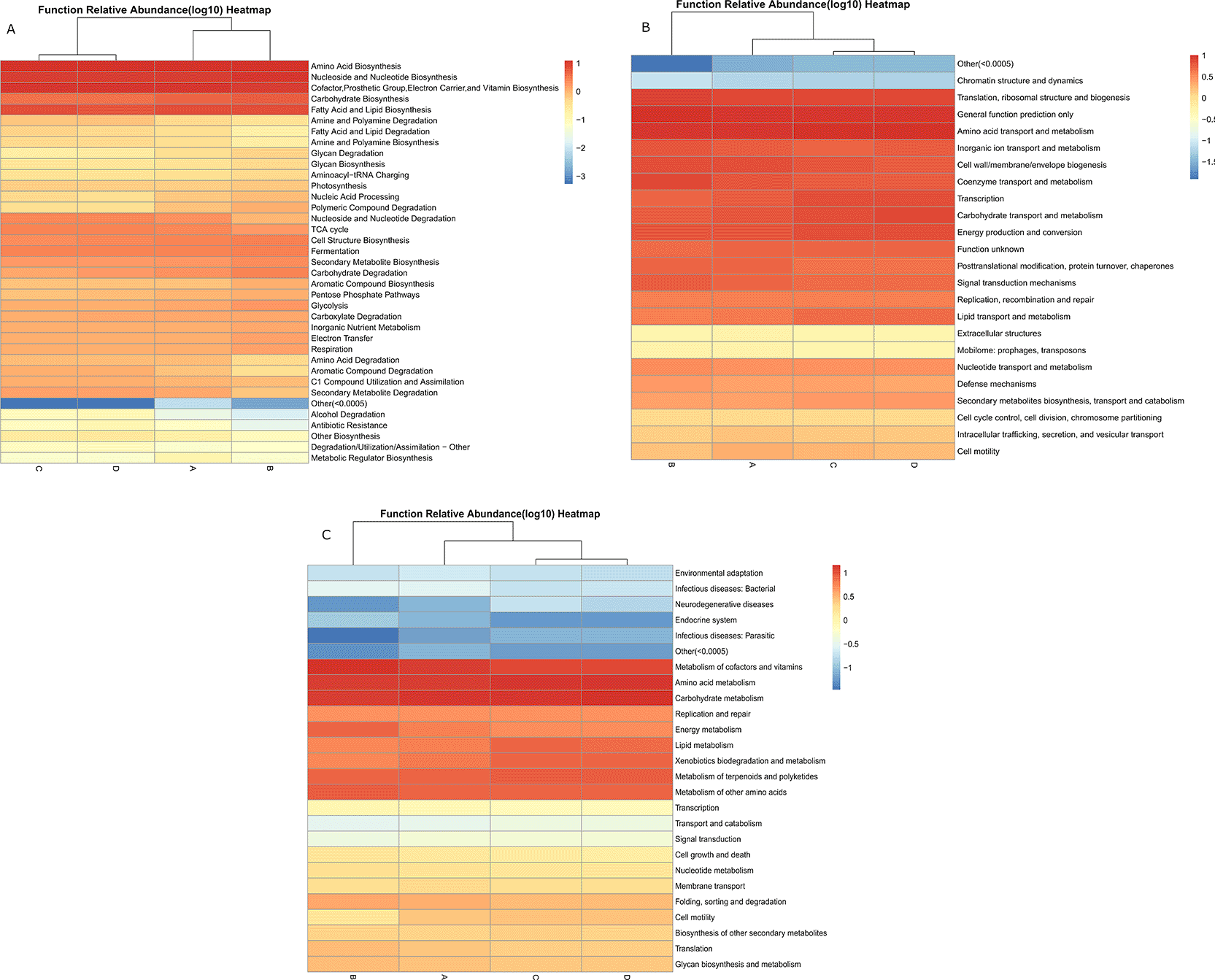
Functional annotations were performed using three databases to predict active metabolic pathways within the analyzed microbial communities: (8-A) MetaCyc pathways, (8-B) COG (Clusters of Orthologous Groups) functional categories, and (8-C) KO (KEGG Orthology) metabolic profiles. The results highlight key pathways associated with amino acid, carbohydrate, and lipid metabolism, as well as secondary metabolite biosynthesis and stress adaptation.
At the MetaCyc level, pathways involved in the biosynthesis of amino acids, carbohydrates, fatty acids, and lipids were the most highly represented across all plant species. Pathways related to secondary metabolite and aromatic compound biosynthesis were also abundant, indicating strong metabolic potential for producing bioactive compounds. In contrast, pathways associated with carboxylate degradation, amino acid degradation, and inorganic nutrient metabolism occurred at moderate abundance, while antibiotic resistance and alcohol degradation were minimally represented. The COG annotation supported these results, showing enrichment in categories related to amino acid, carbohydrate, and lipid transport and metabolism, as well as secondary metabolite biosynthesis, transport, and catabolism. Other dominant functions included energy production and conversion, signal transduction, and defense mechanisms, reflecting the metabolic versatility and environmental responsiveness of the bacterial communities. The KEGG Orthology (KO) functional profiles further confirmed high representation of pathways involved in amino acid and carbohydrate metabolism, followed by xenobiotic biodegradation, terpenoid and polyketide metabolism, and DNA replication and repair. Functions linked to infectious or parasitic diseases, endocrine system, and neurodegenerative pathways appeared at low abundance, indicating that these endophytic communities are primarily oriented toward metabolic adaptation and biosynthesis rather than pathogenicity. These functional predictions suggest that the desert plant endophytes are metabolically active in nutrient cycling, secondary metabolite production, and environmental adaptation, supporting the resilience of their host plants under arid and nutrient-limited desert conditions.
Current projections indicate that desertified land may expand to cover nearly 50% of terrestrial ecosystems by 2050, threatening biodiversity, food security, and soil fertility (Bissenbayeva et al., 2024). A sustainable solution to confront this threat lies in the unique microbial community of the desert environment. These microbes demonstrate remarkable adaptability and stability under extreme abiotic stressors. Understanding their composition, functions, and metabolic patterns is essential to grasp their ecological role and potential contributions to climate adaptation and sustainable agriculture (Alsharif et al., 2020). Recent studies have emphasized that desert microbiomes act as resilience models, providing insights into survival under aridity, salinity, and high temperature (Milli et al., 2024; T. Zhang et al., 2022). Using an eDNA-based approach, this study characterized the endophytic microbial composition of four desert plants native to the Arabian Peninsula: Z. mandavillei, T. zeyheri, L. arabicum, and C. conglomeratus. Among them, L. arabicum and C. conglomeratus exhibited the highest microbial abundance and richness, while T. zeyheri showed the least diversity. To our knowledge, this is the first comparative microbial profiling of these four species, expanding the limited literature on desert endophytes of the Arabian Peninsula.
Our findings revealed that the most abundant bacterial classes across samples were Cyanophyceae, Alphaproteobacteria, Actinomycetia, and Gammaproteobacteria. These groups are commonly reported in arid soils and play critical roles in nutrient cycling, nitrogen fixation, and plant growth promotion (Hood et al., 2017). Many of these taxa exhibit strong tolerance to UV radiation, salinity, and desiccation, traits essential for desert survival (Nimbeshaho et al., 2024; Garcia-Pichel, 2023).
Alphaproteobacteria and Actinomycetia include genera such as Streptomyces, Rhizobium, and Agrobacterium, previously documented for their ability to produce secondary metabolites and antibiotics under extreme stress (Patel et al., 2024). The presence of Gemmatimonadota reflects its known role in phosphorus solubilization, improving nutrient-poor desert soils (Craswell et al., 2021). The Actinomycetota class contributes to nitrogen and phosphorus mobilization by secreting extracellular enzymes that break down complex organic compounds, while also generating bioactive metabolites that inhibit pathogens, thus protecting desert seedlings (Tidimalo et al., 2024). The dominance of Cyanophyceae highlights their dual role in carbon and nitrogen fixation and soil stabilization by producing extracellular polysaccharides that reduce erosion and enhance water retention (Garcia-Pichel, 2023). Alphaproteobacteria further supports plant resilience by synthesizing phytohormones such as auxins and ACC deaminase that improve drought tolerance (Fanai et al., 2024; Yu et al., 2025). Our data showed that Zygophyllum roots were enriched in Cyanophyceae and Proteobacteria, consistent with reports highlighting these taxa as dominant endosphere members in arid ecosystems (Khan et al., 2022). Similarly, Tribulus was enriched in Cyanophyceae and Alphaproteobacteria, aligning with findings in T. terrestris that demonstrated phosphate solubilization and siderophore production as key adaptive mechanisms (Eida et al., 2018).
Functional predictions revealed highly active metabolic profiles characterized by strong biosynthetic and energy-generating pathways, consistent with prior studies (Ronca et al., 2015). Enrichment of pathways involved in amino acid, lipid, and carbohydrate biosynthesis indicates that these microbes maintain strong primary metabolism even under nutrient scarcity. The dominance of post-translational modification, DNA replication, and transcription pathways reflects high metabolic plasticity and environmental resilience (Guisbert et al., 2004). Lipid metabolism, particularly fatty acid biosynthesis, ensures membrane integrity and fluidity under heat stress and provides energy under nutrient limitation, enhancing microbial drought tolerance (Coclet et al., 2022). Similarly, oxidative phosphorylation supports energy generation under fluctuating oxygen and moisture conditions. High carbohydrate metabolism underscores microbial ability to utilize complex carbon sources during scarcity, facilitating plant survival in nutrient-poor soils (Y. Zhang et al., 2025). The elevated expression of amino acid and secondary metabolite pathways reflects the production of stress-protective compounds that contribute to plant–microbe mutualism (Ortiz et al., 2021). Additionally, aromatic compound degradation supports carbon recycling by breaking down complex molecules such as lignin and phenols, thus promoting soil fertility (Wang et al., 2025). Xenobiotic metabolism via cytochrome P450 further enables detoxification of harmful metabolites, safeguarding both plant and microbial communities (Schütz et al., 2021). These findings underscore the metabolic adaptability of desert microbiomes, aligning with recent reports highlighting endophytic microbial resilience to extreme abiotic stress and their potential role in soil stabilization and greenhouse gas modulation (Knight et al., 2024).
Our study highlights the desert microbiome as a model for climate-resilient ecosystems, providing unique microbial solutions to address desertification challenges. These microbial consortia support sustainability through nutrient cycling, plant growth promotion, and carbon fixation (Pointing & Belnap, 2012; Makhalanyane et al., 2015). Their adaptive traits, such as stress-protective pigments, osmolyte production, and efficient water retention, mirror the resilience mechanisms needed for future sustainable agriculture (Bolan et al., 2024). Desert-adapted endophytes could be harnessed as bioinoculants or synthetic microbial communities to improve crop performance in degraded and arid lands. Recent reviews emphasize that inoculation with extremophilic microbes enhances soil fertility, reduces greenhouse gas emissions, and promotes drought tolerance in crops (Soheili Esfahani, 2025; Ali et al., 2025). These microbial ecosystems could therefore contribute directly to climate change mitigation, supporting the United Nations Sustainable Development Goals for food security and environmental conservation. The unique endophytes identified here are candidates for isolating novel genes and metabolites with potential use in biostimulants, biofertilizers, and stress-tolerant crop engineering. Exploring their genomes and metabolites could also lead to drug discovery and environmental biotechnology innovations (Pantigoso et al., 2025).
Mousa et al. (2025). Sequencing data for “Plant Microbiome of the Arabian Peninsula Desert Reveals Unique Structural and Functional Adaptations Supporting Climate Resilience.” NCBI Sequence Read Archive (SRA): BioSample SAMN52919711–SAMN52919714. Available at: https://www.ncbi.nlm.nih.gov/biosample/52919711
All sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under accession numbers SAMN52919711, SAMN52919712, SAMN52919713, SAMN52919714.
https://www.ncbi.nlm.nih.gov/biosample/52919711
https://www.ncbi.nlm.nih.gov/biosample/52919712
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)