Revised
Amendments from Version 1
We thank the reviewers for the helpful suggestions and comments that improved both our work and our manuscript. This new version of the manuscript addresses all the suggestions mentioned by the reviewers and presents a new version of Cyrface, version 2.0. The major differences are: (i) Cyrface is now compatible with Cytoscape 3; (ii) the manuscript was adapted and some parts re-written to address the reviewer’s suggestions; (ii) Rserve is now automatically installed and configured. This greatly simplifies the usage of Cyrface, since it saves the users of having to open R and manually initialising Rserve.
We thank the reviewers for the helpful suggestions and comments that improved both our work and our manuscript. This new version of the manuscript addresses all the suggestions mentioned by the reviewers and presents a new version of Cyrface, version 2.0. The major differences are: (i) Cyrface is now compatible with Cytoscape 3; (ii) the manuscript was adapted and some parts re-written to address the reviewer’s suggestions; (ii) Rserve is now automatically installed and configured. This greatly simplifies the usage of Cyrface, since it saves the users of having to open R and manually initialising Rserve.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Introduction
The availability of high-throughput experimental data has led to the development of multiple computational methods to analyse these data. One of the most used environments is the statistical programming language R1. Multiple R packages for computational biology and bioinformatics are available in various resources such as the Comprehensive R Archive Network (CRAN). Furthermore, Bioconductor2 provides a large collection of curated packages to analyse biological data developed in R. These packages are subject to stringent quality control in terms of functionality and documentation. It is an open-source project hosting 824 active and curated software packages as of May 2014.
For those not familiar with computational programming, learning R and using its packages can be a time consuming task. Therefore, the use of intuitive graphical interfaces that simplifies their use can enhance the usability of these R packages. Cytoscape3,4 is a Java open-source framework with an intuitive graphical interface devoted to the visualization and analysis of networks. It is arguably one of the most used tools in bioinformatics, and has a variety of user developed extensions to solve numerous computational biology problems. These user defined extensions are termed plug-ins (1.x and 2.x) or apps (3.x) depending of which version of Cytoscape is being used.
Here, we present Cyrface, an app for Cytoscape that facilitates an interface between any R package and Cytoscape. Cyrface is designed to integrate the major strengths of R and Cytoscape environments by providing a general Java to R interface. By linking these two environments, Cyrface allows one to use Cytoscape as a graphical user interface for R packages. It also enables Cytoscape apps to access the wealth of methods implemented in R.
Workflow management systems such as Taverna5 and Galaxy6–8 can call R packages from a graphical user interface (GUI)-based interface. Taverna is a standalone Java open-source tool for the general development and execution of workflows. Galaxy is an open-source web-platform to assemble workflows based on genomic experimental data analysis. Thus, Cyrface complements Taverna and Galaxy by enhancing GUIs for R within a different environment with complementary features.
RCytoscape9 is another tool that exists to link R and Cytoscape. It is a Bioconductor R package that establishes a connection between R and Java. The fundamental difference between RCytoscape and Cyrface is that RCytoscape supports the connection from R to Java, whereas Cyrface allows a connection from Java to R. A typical use of RCytoscape would be to handle experimental data from R and transfer the biological network to Cytoscape while controlling it within R. Hence, RCytoscape and Cyrface provide complementary features.
This paper is structured as follows: Firstly, we provide a description of the implementation of Cyrface. Then, to illustrate the usefulness of Cyrface, we show two existing apps, CytoCopteR10 and DrugVsDisease (DvD)11, that make use of Cyrface, and we demonstrate an implementation of a simplified version of the DataRail12 workflow. Finally, we discuss on-going and future developments.
Implementation
Cyrface is a Java open-source framework developed to establish the connection between Cytoscape and R. Interaction between these two different environments (invoking R within Java) is not natively supported by Java. Therefore, to achieve this Cyrface uses the external libraries RCaller13 and Rserve14.
On the one hand, to support the communication between Java and R, RCaller uses an R package called Runiversal that converts the R objects into an XML format, thus allowing the R objects to be read by Java.
On the other hand, Rserve establishes a TCP/IP server allowing other programs from various languages to connect to an R session and access its features. Rserve is currently being used by several mature projects, among them the Taverna workflow management system5.
Rserve and RCaller libraries are integrated in Cyrface by implementing RserveHandler and RCallerHandler Java classes, respectively. Both classes extend the abstract class RHandler that contains the signature of all the necessary methods to establish and maintain a connection with R. Figure 1 depicts the hierarchical structure of these classes and the connection points between these two different environments.
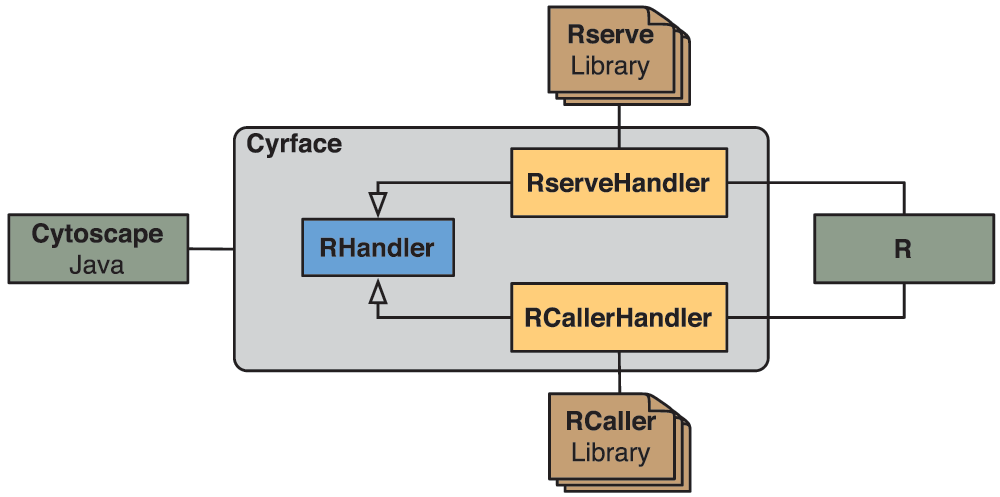
Figure 1. Diagram of the Cyrface interaction layer with R.
Within the grey box the class hierarchy of the classes responsible for establishing the connection between Cytoscape and R is represented. RHandler is an abstract Java class that is extended by RserveHandler and RCallerHandler classes that add support to Rserve and RCaller libraries, respectively. The connection from Java to R can be achieved using either RserveHandler or RCallerHandler classes, or other classes that successfully extend RHandler.
Cyrface software architecture is designed to allow the integration of other Java libraries that facilitate the connection between Java and R. Thereby, this structure allows one to take advantage of particular strengths of different libraries and to adapt to particular requirements of the users. For instance, execute R commands automatically without requiring first to manually initiate an R session.
Cyrface uses Cytoscape’s features, such as the Command Line. The Command Line offers the users the ability to script basic commands in Cytoscape, such as import, display or modify networks through a simple command line or script file. A useful feature of the Command Line is the ability of performing repetitive tasks automatically. By supporting this tool Cyrface extends the possibility of the users to integrate in their scripts methods developed in R together with common Cytoscape features. The Command Tool Dialog window can be used to dynamically execute the necessary R commands. This can be useful, for example when debugging a script.
On Cyrface’s homepage, we provide an illustrative example using the Command Line Dialog tool to plot some features of an existing and publicly available data-set termed, iris13, using the well known plotting library ggplot14. The iris data-set is widely used in the field of pattern recognition and machine learning and is subdivided into different classes, where each class defines the type of the plant iris. This is an illustrative example to demonstrate how Cyrface can within Cytoscape perform any task in the R environment and collect the respective output.
Results and discussion
A typical use of Cyrface is to provide a graphical user interface to R packages within Cytoscape. Cyrface is currently being used by two Cytoscape plug-ins, CytoCopteR10 and DvD11.
CytoCopteR10 provides a simple step-by-step interface allowing users without any experience in R to use the CellNOptR (www.cellnopt.org) package and handle the input and output networks in Cytoscape. CellNOptR is an open-source software package that provides methods for building predictive logic models from signalling networks using experimental measurements of activation of proteins upon perturbation.
DvD11, Drug vs Disease, is an R package that provides a workflow for the comparison of drug and disease gene expression profiles. It provides dynamic access to databases, such as Array Express15, to compare drug and disease signatures to generate hypotheses of drug-repurposing.
CytoCopteR and DvD are two examples of how Cyrface captures the strengths of both environments. On one side, R provides a wealth of bioinformatics and biostatistics packages with very comprehensive resources such as Bioconductor and CRAN. On the other side, Cytoscape facilitates a user-friendly graphical interface for network visualisation and analysis, complemented with a variety of plug-ins or apps addressing different computational biological problems. Cyrface links these two environments by providing a way to develop user-friendly graphical interfaces for R packages by embedding them within Cytoscape.
As another illustrative example, we implemented in Cyrface a simplified version of the DataRail12 workflow. This example is designed to illustrate how one can use methods already available in R and build a graphical user interface in Cytoscape to access them.
DataRail is an open-source MATLAB toolbox that handles experimental data in a tabular format and provides methods to maximize and extract information using internal or external tools. Experimental data is stored in a format termed Minimum Information for Data Analysis in Systems Biology (MIDAS). This is a tabular format that specifies the layout of experimental data files12. A typical use of DataRail is to import, store and process the input information from instruments using the MIDAS format, and export it to other MIDAS compliant software, such as CellNOptR.
The DataRail workflow implemented in Cyrface is structured in several sequential steps that allows the users to import, normalise and visualise experimental data-sets stored in the MIDAS format (Figure 2). The workflow is tested using an in silico generated data-set and a signalling network from16.
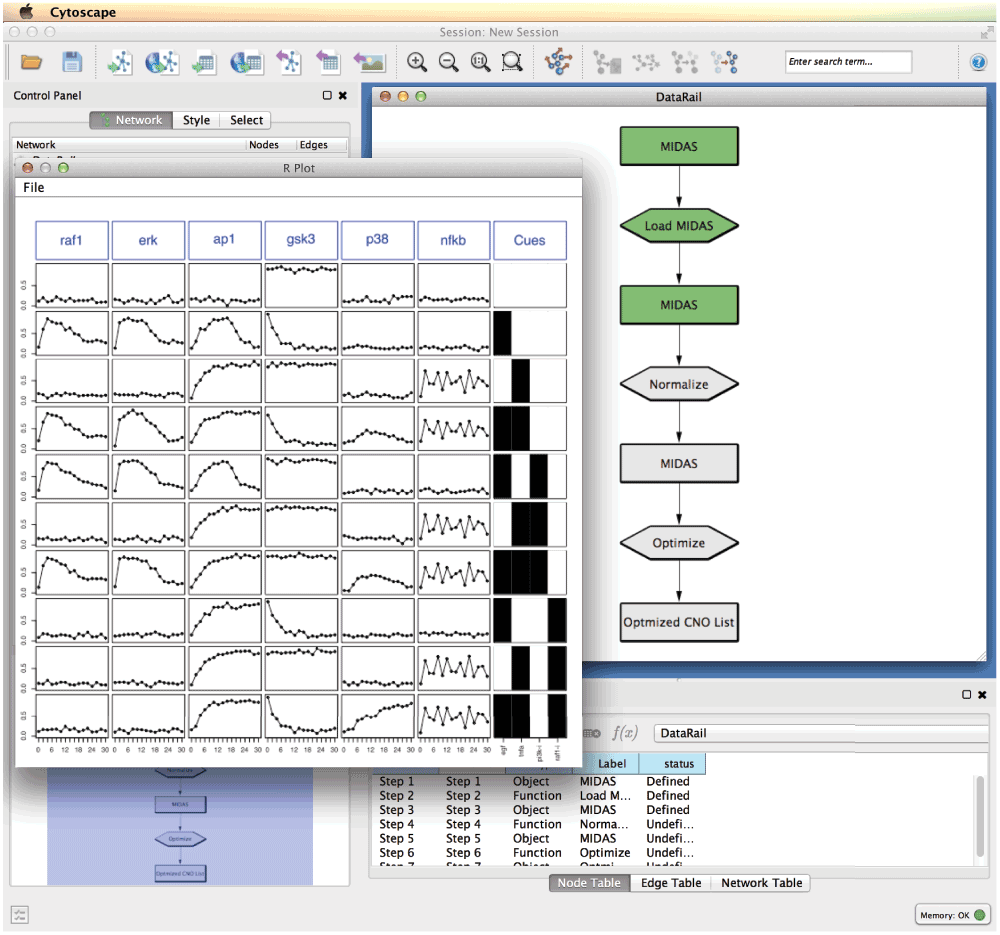
Figure 2. The Cyrface implementation of the DataRail12 workflow.
The rounded rectangles represent the MIDAS files containing the experimental data at a given state. Hexagon nodes represent functions such as load or normalise. Green identifies steps that were successfully executed and grey identifies those that were not run yet. The data-set shown represents the normalised values of the protein activity state of a set of proteins (columns) under different stimulatory conditions (rows).
An extension to the workflow was subsequently added to support the model training function form CellNOptR package10. CellNOptR training function maximises the fit of the experimental data and the corresponding prior-knowledge network, by generating and optimising a logic model. Thereby, through an intuitive graphical interface, users are able to select a biological network and use it to assess the quality of the fit with a corresponding data set of experimental data. This extension illustrates how one can in principle embed any R package in such a workflow, but it does not replace the CytoCopteR app as a complete interface for CellNOptR.
The workflow supports the SIF network format, which is supported by Cytoscape, but also the Systems Biology Markup Language Qualitative Models (SBML Qual) format16. SBML Qual is an extension of the SBML format and is proposed to provide a standard representation for logic and qualitative models of biological networks. Support for importing models stored in SBML Qual format is achieved using the jSBML library17 and the respective SBML Qual package. Supplementary material 1 provides a step-by-step tutorial on how to use the workflow.
Conclusions
Here, we present Cyrface; a bioinformatics Java library that provides a general interaction between Cytoscape and R. Cyrface offers a way to combine a friendly graphical interface within the Cytoscape environment with any R package. A GUI should benefit beginners and occasional users; as well as being useful for training and illustration purposes, it extends the accessibility of the tool to those not familiar with the R command line interface.
Moreover, Cyrface complements other libraries such as Rserve since, (i) it is capable of using Rserve, RCaller or any other existing Java library to query R, and (ii) it provides a tailored implementation for Cytoscape, providing interfaces that are suited to Cytoscape features, such as the support of the Command Dialog tool.
Cyrface’s homepage (see Software Details section) contains the link to download Cyrface and user-guide instructions. A few examples demonstrating the usefulness of the tool and the different supported libraries are also shown and explained. The source-code of Cyrface is publicly available on its GitHub webpage (see Software Details section).
Future features for Cyrface will include providing connections to Cytoscape.js, improvements to the DataRail workflow and further developing and testing future features, such as add support access to remote servers of Rserve.
A common scenario in an interdisciplinary field such as network biology, is one where there is on one side an expert on a certain biological question, who has data to address this question and, on the other side a computational scientist who develops algorithms, but is less familiar with the experiments. To help to bridge this situation tools like Cyrface facilitate to encapsulate sophisticated algorithms developed in R in a user-friendly interface within the Cytoscape framework, to enable non-experts to apply these algorithms.
Software details
Homepage: http://www.ebi.ac.uk/saezrodriguez/cyrface/
Software available from: http://apps.cytoscape.org/apps/cyrface
Latest source code: https://github.com/EmanuelGoncalves/cyrface
Source code as at the time of publication: https://github.com/F1000Research/cyrface
Archived source code as at the time of publication: http://www.dx.doi.org/10.5281/zenodo.1015318
License: GNU General Public License version 3.0 (GPLv3)
Author contributions
JSR initiated and guided the project. EG designed the software architecture. EG and FM implemented Cyrface. EG and JSR prepared the manuscript.
Competing interests
No competing interests were disclosed.
Grant information
We acknowledge with thanks the financial support from the EU through the project “BioPreDyn” (ECFP7-KBBE-2011-5 Grant number 289434).
Acknowledgements
The authors would like to thank Martijn Van Iersel for helpful discussions and suggestions in the earlier stages of Cyrface development.
Supplementary materials
Supplementary material 1: Cyrface DataRail workflow tutorial.
Cyrface Manual
Cyrface is a bioinformatics Java library that provides a general interaction between Cytoscape and R. Cyrface offers a way to combine a friendly graphical interface within the Cytoscape environment with any R package. A GUI should benefit beginners and occasional users; as well as being useful for training and illustration purposes, it extends the accessibility of the tool to those not familiar with the R command line interface.
This tutorial is intended for Cyrface v2.0 that requires Cytoscape v3.1 and R 3.x. The following materials are all available online on Cyrface homepage: http://www.ebi.ac.uk/saezrodriguez/cyrface
Software Requirements
Please be sure that you have the following software installed and working:
It is very important that Cytoscape version is equal or greater than 3.1.0
Linux users must run the following commands before using Cytocopter
Introduction
http://www.ebi.ac.uk/saezrodriguez/cyrface
Cyrface1 establishes an interface between R and Cytoscape2,3 by using different Java-R libraries, e.g. Rserve, RCaller. Cyrface can be used as a Cytoscape App, e.g. to run R commands within Cytoscape, or used as a library to allow your App to connect to R.
This is developed under a GNU open-source license and the source code can be accessed from the respective GitHub project.
Installation
Install from Cytoscape
Click Apps on Cytoscape top bar followed by Apps Manager menu then search for Cyrface and click install. This may take a few minutes.
Install from file
Download the jar file of Cyrface from the following URL:
Click Apps on Cytoscape top bar followed by Apps Manager then click Install from file… button and search Cyrface jar file.
Install manually
Cyrface can also be installed manually by copying the jar file mentioned before to CytoscapeConfiguration folder. CytoscapeConfiguration folder is kept in the user home folder. Drag the App jar file into the following folder:
After moving the jar file start Cytoscape.

Cyrface DataRail Tutorial
Introduction
This tutorial assumes that Cytoscape as well as R is already installed. The necessary files for this tutorial is a network file in SBML-Qual format and the corresponding experimental data in MIDAS format.
Study case
To illustrate the usefulness of Cyrface we will use a simple implementation of the DataRail workflow4.
The accompanying in silico data (MIDAS file format4) replicates biologically plausible behaviour that has been seen in intracellular signalling networks, such as the transient behaviour of ERK activation and the oscillatory dynamics of NFkB translocation from the cytoplasm to the nucleus.
MIDAS experimental data format
The MIDAS format (Minimum Information for Data Analysis in Systems Biology)4 is a comma-separated file that specifies the layout of experimental data files.
Each row represents a single experimental sample; each column represents one sample attribute, such as treatment condition, or value obtained from an experimental assay.
SBML-Qual network format
SBML-Qual format5 is an extension to the System Biology Markup Language (SBML) for Qualitative Models (Qual). In one sentence, it is designed to provide a standard mean for the exchange of logical models or regulatory and signalling networks.
For more details regarding the specifications please see5.
Tutorial
1. To start Cyrface’s DataRail workflow go to Apps -> Cyrface -> DataRail
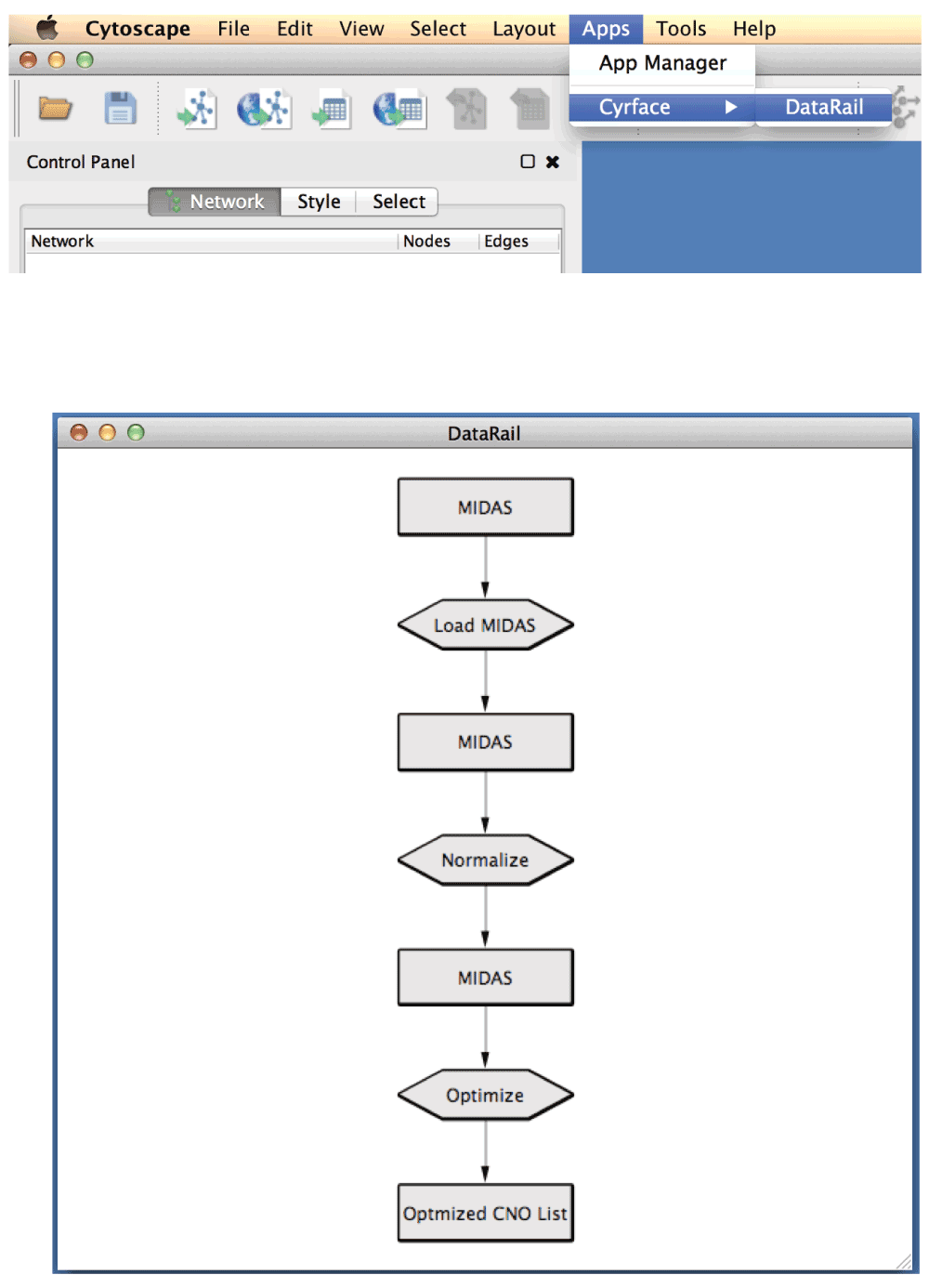
2. The full workflow should now be visible
3. Right-click on the top MIDAS node and then select Cyrface -> Set MIDAS file… to select the desired MIDAS file. After the MIDAS file is selected the node should turn green.
Raw data MIDAS file:
www.ebi.ac.uk/saezrodriguez/cyrface/resources/ToyDataPB10raw.csv
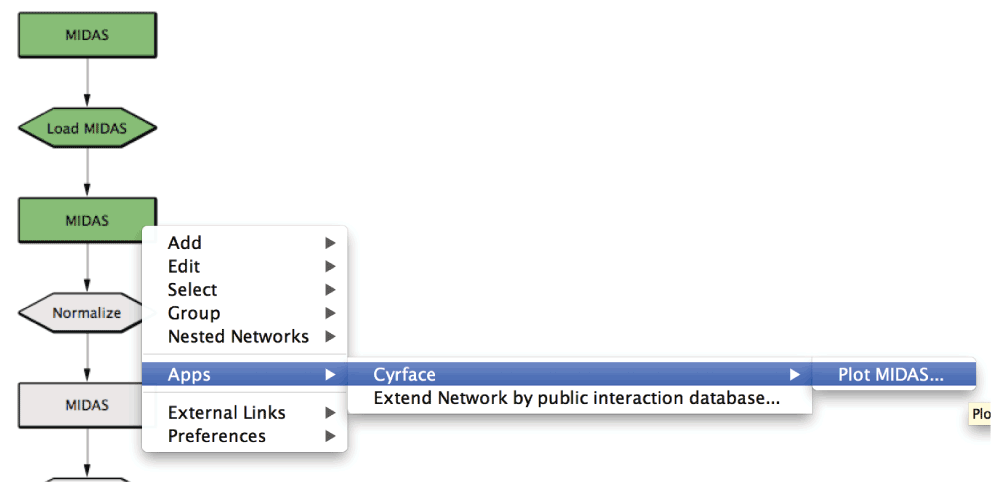
4. Right-click on Load MIDAS node and then select Cyrface -> Load MIDAS… option to load the previously selected MIDAS file. After the file is loaded the node should turn green.
5. After the MIDAS file is successfully loaded the second MIDAS node is now green showing that it’s ready to be normalized or visualized.
6. Right-click on the respective MIDAS node and the selecting the Cyrface -> Plot MIDAS… option will pop-up a plot of the data
7. Right-click on the Normalize node to run the normalization function. A pop-up window will show up to allow the user to define the Normalization function arguments:
a.
EC50Data: parameter for the scaling of the data between 0 and 1, default=0.5
b.
Detection: minimum detection level of the instrument, everything smaller will be treated as noise (NA), default to 0
c.
Saturation: saturation level of the instrument, everything over this will be treated as NA, default to Inf
8. After normalizing the MIDAS file it can be plotted as previously and/or exported.

Optional steps
Cyrface’s DataRail4 Workflow is also linked to the CellNOptR6 R package allowing the users to optimize a selected prior knowledge network against the just normalized MIDAS file.
9. Right-click on the Optimize node and select Cyrface -> Optimize… function will pop-up a file browser to select the model file. Both SIF and SBML-Qual5 formats are supported.
10. The optimization may take awhile and it’s executed using the defaults values defined in CellNOptR
11. Right-click on the Optimized CNO List will show how well the optimized model fit the data.
12. For more details about the normalization function and the optimization method please visit CellNOptR package in Bioconductor or CellNOpt homepage

References
1. Gonçalves, E. & Saez-Rodriguez, J. Cyrface: An interface from Cytoscape to R that provides a user interface to R packages. F1000Res
2, 192 (2013).
2. Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research
13, 2498–2504 (2003).
3. Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L. & Ideker, T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics
27, 431–432 (2011).
4. Saez-Rodriguez, J. et al. Flexible informatics for linking experimental data to mathematical models via DataRail. Bioinformatics
24, 840–847 (2008).
5. Chaouiya, C. et al. SBML qualitative models: a model representation format and infrastructure to foster interactions between qualitative modelling formalisms and tools. BMC Syst Biol
7, 135 (2013).
6. Terfve, C. et al. CellNOptR: a flexible toolkit to train protein signaling networks to data using multiple logic formalisms. BMC Syst Biol
6, 133 (2012).
Faculty Opinions recommendedReferences
- 1.
Team RC: R: A Language and Environment for Statistical Computing. GNU GLP. Reference Source
- 2.
Gentleman RC, Carey VJ, Bates DM, et al.:
Bioconductor: open software development for computational biology and bioinformatics.
Genome Biol.
2004; 5(10): R80. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 3.
Shannon P, Markiel A, Ozier O, et al.:
Cytoscape: a software environment for integrated models of biomolecular interaction networks.
Genome Res.
2003; 13(11): 2498–2504. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 4.
Smoot ME, Ono K, Ruscheinski J, et al.:
Cytoscape 2.8: new features for data integration and network visualization.
Bioinformatics.
2011; 27(3): 431–432. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 5.
Wolstencroft K, Haines R, Fellows D, et al.:
The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, web or in the cloud.
Nucleic Acids Res.
2013; 41(Web Server issue): W557–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 6.
Goecks J, Nekrutenko A, Taylor J, et al.:
Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences.
Genome Biol.
2010; 11(8): R86. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
Blankenberg D, Von Kuster G, Coraor N, et al.:
Galaxy: a web-based genome analysis tool for experimentalists.
Curr Protoc Mol Biol.
2010; Chapter 19(Unit 19.10): 19.10.1–21. PubMed Abstract
| Publisher Full Text
- 8.
Giardine B, Riemer C, Hardison RC, et al.:
Galaxy: a platform for interactive large-scale genome analysis.
Genome Res.
2005; 15(10): 1451–1455. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 9.
Shannon PT, Grimes M, Kutlu B, et al.:
RCytoscape: tools for exploratory network analysis.
BMC Bioinformatics.
2013; 14: 217. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 10.
Terfve C, Cokelaer T, Henriques D, et al.:
CellNOptR: a flexible toolkit to train protein signaling networks to data using multiple logic formalisms.
BMC Syst Biol.
2012; 6: 133. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Pacini C, Iorio F, Gonçalves E, et al.:
DvD: An R/Cytoscape pipeline for drug repurposing using public repositories of gene expression data.
Bioinformatics.
2013; 29(1): 132–134. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 12.
Saez-Rodriguez J, Goldsipe A, Muhlich J, et al.:
Flexible informatics for linking experimental data to mathematical models via DataRail.
Bioinformatics.
2008; 24(6): 840–847. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
RCaller. Reference Source
- 14.
Urbanek S:
A Fast Way to Provide R Functionality to Applications. 2003; 2. . Reference Source
- 15.
Rustici G, Kolesnikov N, Brandizi M, et al.:
ArrayExpress update--trends in database growth and links to data analysis tools.
Nucleic Acids Res.
2013; 41(Database issue): D987–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 16.
Chaouiya C, Berenguier D, Keating SM, et al.:
SBML Qualitative Models: a model representation format and infrastructure to foster interactions between qualitative modelling formalisms and tools.
BMC Syst Biol.
2013; 7: 135. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 17.
Drager A, Rodriguez N, Dumousseau M, et al.:
JSBML: a flexible Java library for working with SBML.
Bioinformatics.
2011; 27(15): 2167–2168. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 18.
Gonçalves E, Mirlach F, Saez-Rodriguez J:
F1000Research/cyrface.
ZENODO.
2014. Data Source
Comments on this article Comments (0)