Gonçalves E and Saez-Rodriguez J. Cyrface: An interface from Cytoscape to R that provides a user interface to R packages [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2013, 2:192 (https://doi.org/10.12688/f1000research.2-192.v1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
This article is included in the Bioconductor gateway.
This article is included in the EMBL-EBI collection.
Abstract
There
is an increasing number of software packages to analyse biological experimental
data in the R environment. In
particular, Bioconductor, a
repository of curated R packages, is one of the most comprehensive resources for
bioinformatics and biostatistics. The use of these packages is increasing, but
it requires a basic understanding of the R language, as well as the syntax of
the specific package used. The availability of user graphical interfaces for
these packages would decrease the learning curve and broaden their application.
Here,
we present a Cytoscape plug-in termed
Cyrface that allows Cytoscape plug-ins to connect to any function
and package developed in R. Cyrface can be used to run R packages from within the Cytoscape environment making use of a
graphical user interface. Moreover, it links the R packages with the
capabilities of Cytoscape and its plug-ins, in particular network visualization
and analysis. Cyrface’s utility has been demonstrated for two Bioconductor
packages (CellNOptR and DrugVsDisease), and here we further illustrate its
usage by implementing a workflow of data analysis and visualization. Download
links, installation instructions and user guides can be accessed from the Cyrface homepage (http://www.ebi.ac.uk/saezrodriguez/cyrface/).
Corresponding author:
Julio Saez-Rodriguez
Competing interests:
No competing interests were disclosed.
Grant information:
We acknowledge with thanks the financial support from the EU through the project “BioPreDyn” (ECFP7-KBBE-2011-5 Grant number 289434).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The availability of high-throughput experimental data has led to the development of multiple computational methods to analyse these data. Arguably, one of the most used environments is the statistical programming language R1. Multiple R packages for computational biology and bioinformatics are available in various resources such as the Comprehensive R Archive Network (CRAN). Furthermore, Bioconductor2 provides a comprehensive collection of packages to analyse biological data developed in R. These packages are subject to stringent quality control in terms of functionality and documentation. It is an open-source project hosting 671 active and curated software packages as of September 2013.
For those not familiar with computational programming, learning R and running packages can be a time consuming task and therefore the use of intuitive graphical interfaces can enhance the usability of the tool. Cytoscape3,4 is a Java open-source framework with an intuitive graphical interface devoted to the visualization and analysis of networks. It is arguably one of the most used tools in bioinformatics, and has a variety of plug-ins to solve numerous computational biology problems. Therefore, we developed Cyrface, a plug-in for Cytoscape that facilitates an interface between any R package and Cytoscape. Cyrface is designed to integrate the major strengths of R and Cytoscape environments by providing a general Java to R interface. By linking these two environments, Cyrface allows one to use Cytoscape as a user interface for R packages and Cytoscape plug-ins in order to reach the wealth of methods implemented in R.
Workflow management systems such as Taverna5 and Galaxy6–8 can call R packages from a graphical user interface (GUI)-based interface. Taverna is a standalone Java open-source tool for the general development and execution of workflows. Galaxy is an open-source web-platform to assemble workflows based on genomic experimental data analysis. Thus, Cyrface complements Taverna and Galaxy by enhancing GUIs for R within a different environment with complementary features.
RCytoscape9 is another tool that exists to link R and Cytoscape. It is a Bioconductor R package that establishes a connection between R and Java in the opposite direction of Cyrface: it supports the connection from R to Java, whereas Cyrface allows a connection from Java to R. A typical use of RCytoscape is to handle experimental data from R and transfer the biological network to Cytoscape while controlling it within R. Hence, RCytoscape and Cyrface provide complementary features.
This paper is structured as follows: Firstly, we provide a description of the implementation of Cyrface. Then, to illustrate the applicability of Cyrface, we show two existing packages, CytoCopteR10 and DrugVsDisease (DvD)11, that make use of Cyrface, and we create a simplified version of the DataRail12 workflow to process and visualize experimental data using methods available in R. Finally, we discuss on-going and future developments.
Implementation
Cyrface is a Java open-source framework developed to establish the connection between Cytoscape and R. Interaction between these two different environments (invoking R within Java) is not natively supported by Java. Therefore, to achieve this Cyrface uses the external libraries RCaller (https://code.google.com/p/rcaller/) and Rserve (http://www.rforge.net/Rserve/).
On the one hand, to support the communication between Java and R, RCaller uses an R package called Runiversal that converts the R objects into an XML format, thus allowing the R objects to be read by Java.
On the other hand, Rserve establishes a TCP/IP server allowing other programs from various languages to connect to an R session and access its features. Rserve is currently being used by several mature projects, among them the Taverna workflow management system5.
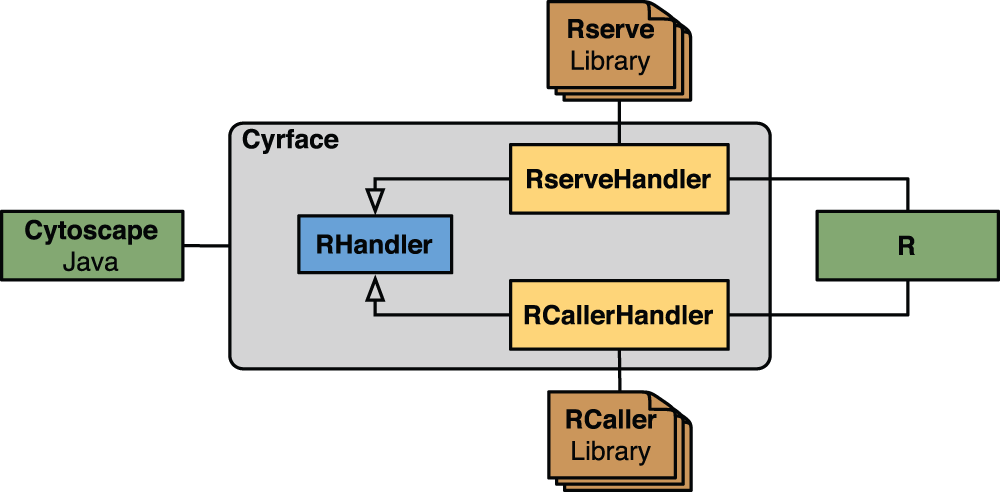
Support for Rserve and RCaller libraries in Cyrface is implemented by the RserveHandler and RCallerHandler Java classes, respectively. Both classes extend the abstract class RHandler that contains the signature of all the necessary methods to establish and maintain a connection with R. Figure 1 depicts the hierarchical structure of the Java classes responsible for handling the connection between Java and R. Moreover, it depicts the connection points between these two different environments.
Figure 1. Diagram of the Cyrface interaction layer with R.
Within the grey box the class hierarchy of the classes responsible for establishing the connection between Cytoscape and R is represented. RHandler is an abstract Java class that is extended by RserveHandler and RCallerHandler classes that add support to Rserve and RCaller libraries, respectively. The connection from Java to R can be achieved using either RserveHandler or RCallerHandler classes, or other classes that successfully extend RHandler.
Cyrface software architecture can be extended to support other Java libraries that facilitate the connection between Java and R. Thereby, this structure allows one to take advantage of particular strengths of different libraries and to adapt to particular requirements of the users, for instance execute R commands automatically without requiring first to manually initiate an R session.
Cyrface uses another Cytoscape plug-in termed CommandTool. CommandTool offers the users the ability to script basic commands in Cytoscape, such as import, display or modify networks through a simple command line. The integration allows the users to use the simple command line of CommandTool to execute R commands within Cytoscape and visualise directly the output. On Cyrface’s homepage (http://www.ebi.ac.uk/saezrodriguez/cyrface/) we provide an example using the CommandTool console to plot several characteristics of the iris data set using the ggplot13 plotting library.
Results and discussion
A typical use of Cyrface is to provide a graphical user interface to R packages within Cytoscape. Cyrface is currently being used by two Cytoscape plug-ins, CytoCopteR10 and DvD11.
CytoCopteR10 provides a simple step-by-step interface allowing users without any experience in R to use the CellNOptR (www.cellnopt.org) package and handle the input and output networks in Cytoscape. CellNOptR is an open-source software package that provides methods for building predictive logic models from signalling networks using experimental measurements.
DvD11, Drug vs. Disease, is an R package that provides a workflow for the comparison of drug and disease gene expression profiles. It provides dynamic access to databases, such as Array Express14, to compare drug and disease signatures to generate hypotheses of drug-repurposing.
The packages mentioned above are two examples of the usefulness of Cyrface in capturing the strengths of two environments. On one side, R provides a wealth of bioinformatics and biostatistics packages with very comprehensive resources such as Bioconductor and CRAN. On the other side, Cytoscape facilitates a user-friendly graphical interface for network visualisation and analysis, complemented with a variety of plug-ins addressing different computational biological problems. Cyrface links these two environments by providing a way to develop user-friendly interfaces for R packages by embedding them within Cytoscape.
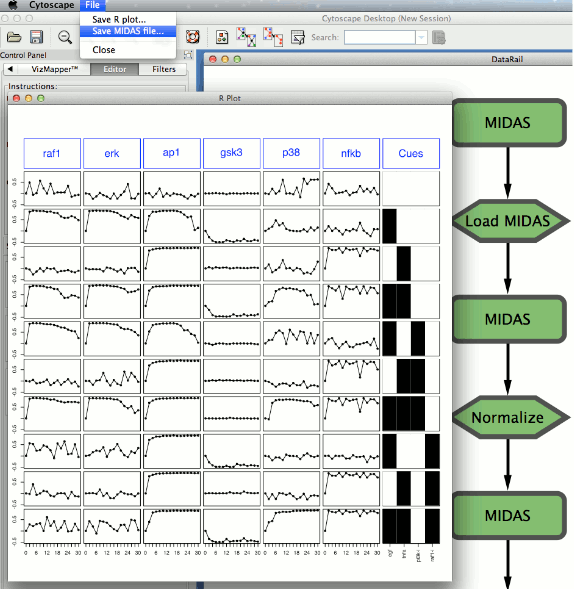
As an illustrative example, Cyrface provides a simple version of the DataRail12 workflow using methods implemented in R. DataRail is an open-source MATLAB toolbox that handles experimental data in a tabular format and provides methods to maximize and extract information using internal or external tools. Saez-Rodriguez et al.12 also proposed an experimental data storing format termed Minimum Information for Data Analysis in Systems Biology (MIDAS).This is a tabular format based upon the minimum-information standards that specifies the layout of experimental data files. A typical use of DataRail is to import, store and process the input information from instruments using the MIDAS format, and export it to other MIDAS compliant software.
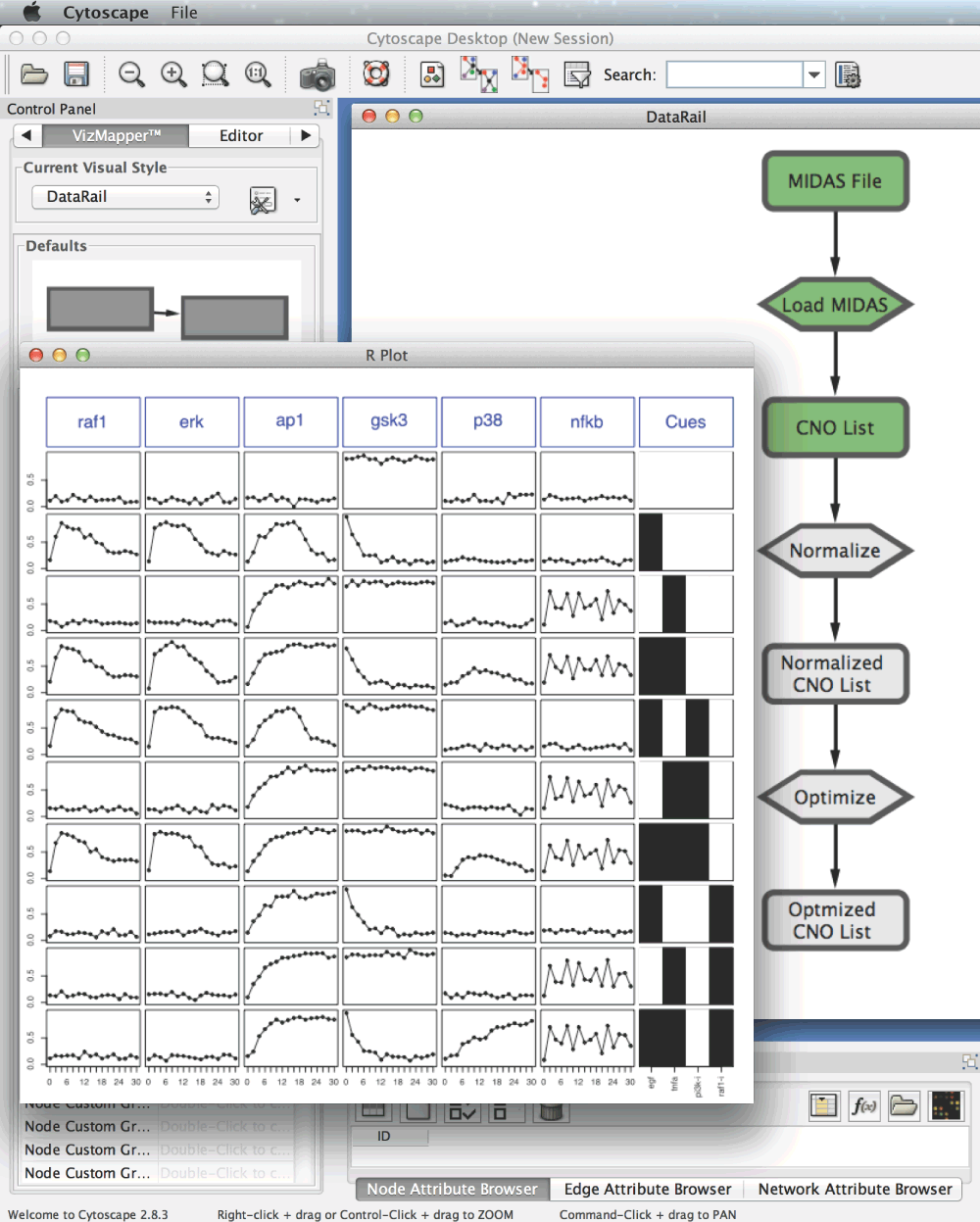
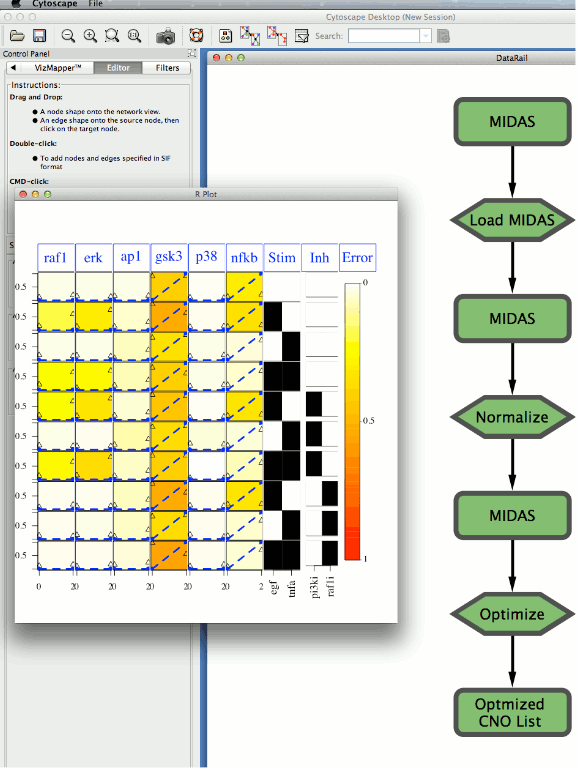
The simplified version of the DataRail workflow implemented in Cyrface is structured in several sequential steps that allows the users to import, normalise and visualise experimental data-sets stored in the MIDAS format (see Figure 2). At any stage the users are able to export and visualise the transformed data set.
Figure 2. The Cyrface implementation of the DataRail12 workflow.
The rounded rectangles represent the MIDAS files containing the experimental data at a given state. Hexagon nodes represent functions such as load or normalise. Green identifies steps that were successfully executed and grey identifies those that were not run yet.
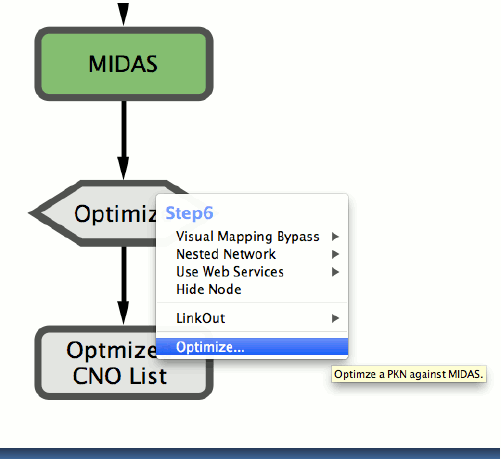
An extension to the workflow was subsequently added to support the CellNOptR10 model training function. CellNOptR uses the experimental data and a corresponding prior-knowledge network to generate a logic model and train it to maximise the fit with the experimental measurements. Thereby, through an intuitive graphical interface, users are able to visualise a biological network, modify it and use it to assess the quality of the fit with a corresponding data set of experimental data.
The workflow supports any network format that is supported by Cytoscape, for example the SIF format. Moreover, the workflow was extended to support the Systems Biology Markup Language (SBML) Qualitative Models (Qual) format15. SBML Qual is an extension of the SBML level 3 standard and is proposed to provide a standard representation for logic and qualitative models of biological networks. The latest specification document for SBML Qual can be found on the package homepage (http://sbml.org/Documents/Specifications/SBML_Level_3/Packages/Qualitative_Models_(qual)). Support for importing models stored in SBML Qual format is achieved using the jSBML library16 and the respective SBML Qual package. Supplementary material 1 provides a step-by-step tutorial and an example on how to use the workflow.
Conclusions
Here, we present Cyrface; a bioinformatics Java library that provides a general interaction between Cytoscape and R. Cyrface offers a way to combine a friendly graphical interface within the Cytoscape environment with any R package. A GUI should benefit beginners and occasional users; as well as being useful for training and illustration purposes, it extends the accessibility of the tool to those not familiar with the R command line interface.
The Cyrface homepage (http://www.ebi.ac.uk/saezrodriguez/cyrface/) contains the link to download Cyrface, and installation and user-guide instructions. A few examples demonstrating the usefulness of the tool and the different supported libraries are also shown and explained. The source-code of Cyrface is publicly available on its Sourceforge webpage (https://sourceforge.net/projects/cyrface/) and permanently available on 10.5281/zenodo.7096.
Future features for Cyrface will include the extension to the new version of Cytoscape, Cytoscape 3, and improvements to the DataRail workflow. These will include increasing its modularity and supporting other features, such as cutting and selecting specific regions of the data.
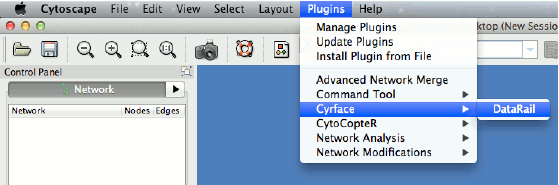
1. To start Cyrface’s DataRail workflow go to “Plugin -> Cyrface -> DataRail”
2. The full workflow should now be visible.
3. Right-click on the top MIDAS node and then select “Set MIDAS file…” to select the desired MIDAS file. After the MIDAS file is selected the node should turn green.
4. Right-click on “Load MIDAS” and then select “Load MIDAS…” option to load the previously selected MIDAS file. After the file is loaded the node should turn green.
5. After the MIDAS file is successfully loaded the second MIDAS node is now green showing that it’s ready to be normalized or visualized.
6. Right-click on the respective MIDAS node and the selecting the “Plot MIDAS…” option will pop-up a plot of the data (the plot can be exported following “File -> Save R plot…”
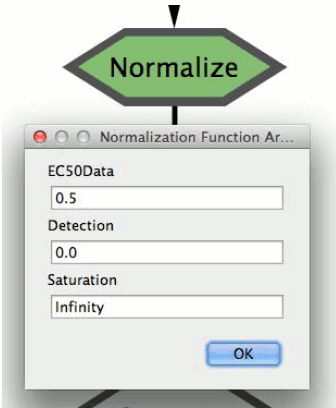
7. Right-click on the “Normalize” node to run the normalization function. A pop-up window will show up to allow the user to define the Normalization function arguments:
a. EC50Data:parameter for the scaling of the data between 0 and 1, default=0.5
b. Detection:minimum detection level of the instrument, everything smaller will be treated as noise (NA), default to 0
c. Saturation:saturation level of the instrument, everything over this will be treated as NA, default to Inf
8. After normalizing the MIDAS file it can be ploted as previously and/or exported.
-- Optional –
Cyrface’s DataRail Workflow is also linked to the CellNOptR R package allowing the users to optimize a selected prior knowledge network against the just normalized MIDAS file.
9. Right-click on the “Optimize” node and select “Optimize…” function will pop-up a file browser to select the model file. Both Sif and SBML-qual formats are supported.
10. The optimization may take awhile and it’s executed using the defaults values defined in CellNOptR
11. Right-click on the “Optimized CNO List” will show how well the optimized model fit the data.
12. For more details about the normalization function and the optimization method please visit CellNOptR package in Bioconductor or CellNOpt homepage
1.
Team RC:
R: A Language and Environment for Statistical Computing.: GNU GLP. Reference Source
2.
Gentleman RC, Carey VJ, Bates DM, et al.:
Bioconductor: open software development for computational biology and bioinformatics.
Genome Biol.
2004; 5(10): R80. PubMed Abstract
| Publisher Full Text
| Free Full Text
4.
Smoot ME, Ono K, Ruscheinski J, et al.:
Cytoscape 2.8: new features for data integration and network visualization.
Bioinformatics.
2011; 27(3): 431–432. PubMed Abstract
| Publisher Full Text
| Free Full Text
5.
Wolstencroft K, Haines R, Fellows D, et al.:
The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, web or in the cloud.
Nucleic Acids Res.
2013; 41(Web Server issue): W557–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
6.
Goecks J, Nekrutenko A, Taylor J, et al.:
Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences.
Genome Biol.
2010; 11(8): R86. PubMed Abstract
| Publisher Full Text
| Free Full Text
7.
Blankenberg D, Kuster Von G, Coraor N, et al.:
Galaxy: a web-based genome analysis tool for experimentalists.
Curr Protoc Mol Biol.
2010; Chapter 19(Unit 19.10): 10.1–21. PubMed Abstract
| Publisher Full Text
10.
Terfve C, Cokelaer T, Henriques D, et al.:
CellNOptR: a flexible toolkit to train protein signaling networks to data using multiple logic formalisms.
BMC Syst Biol.
2012; 6: 133. PubMed Abstract
| Publisher Full Text
| Free Full Text
11.
Pacini C, Iorio F, Gonçalves E, et al.:
DvD: An R/Cytoscape pipeline for drug repurposing using public repositories of gene expression data.
Bioinformatics.
2013; 29(1): 132–134. PubMed Abstract
| Publisher Full Text
| Free Full Text
12.
Saez-Rodriguez J, Goldsipe A, Muhlich J, et al.:
Flexible informatics for linking experimental data to mathematical models via DataRail.
Bioinformatics.
2008; 24(6): 840–847. PubMed Abstract
| Publisher Full Text
| Free Full Text
13.
Wickham H:
ggplot2: Elegant Graphics for Data Analysis. Springer Publishing Company, Incorporated; 2009. Reference Source
14.
Rustici G, Kolesnikov N, Brandizi M, et al.:
Array Express update--trends in database growth and links to data analysis tools.
Nucleic Acids Res.
2013; 41(Database issue): D987–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Chaouiya C, Berenguier D, Keating SM, et al.:
SBML Qualitative Models: a model representation format and infrastructure to foster interactions between qualitative modelling formalisms and tools. arxiv-web3.library.cornell.edu.. Reference Source
We acknowledge with thanks the financial support from the EU through the project “BioPreDyn” (ECFP7-KBBE-2011-5 Grant number 289434).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Gonçalves E and Saez-Rodriguez J. Cyrface: An interface from Cytoscape to R that provides a user interface to R packages [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2013, 2:192 (https://doi.org/10.12688/f1000research.2-192.v1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Shannon PT. Reviewer Report For: Cyrface: An interface from Cytoscape to R that provides a user interface to R packages [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2013, 2:192 (https://doi.org/10.5256/f1000research.2371.r1863)
Cyrface is a welcome addition to the Cytoscape ecosystem; nicely complementary to RCytoscape. My only reservation is one which applies to my own work (the aforementioned RCytoscape), indeed as well as all parts of the Cytoscape ecosystem.
My reservation has two parts:
First:
... Continue reading
Cyrface is a welcome addition to the Cytoscape ecosystem; nicely complementary to RCytoscape. My only reservation is one which applies to my own work (the aforementioned RCytoscape), indeed as well as all parts of the Cytoscape ecosystem.
My reservation has two parts:
First: network biology is in its infancy and as such experimental data are woefully incomplete. Molecular interactions are stochastic, contingent and often very short-lived yet it is exactly these molecular interactions that we need to understand in order to predict and control cellular activity in health and disease.
Second: it is (and may remain) unusual to find researchers, much less clinicians, who are adept at both programming and biomedicine. These two disciplines seem to select for, and then reinforce, different styles of thinking. Therefore progress in this field (call it network biology, or systems biology, or integrative biomedicine) requires hybrid teams: some who are very strong in biological and/or clinical sensibilities and some who are strong in computation and data analysis. Such a hybrid team, at its best, stays together long enough for mutual understanding and communication to emerge, as in the "trading language" which emerged in the world of particle physics in and around linear accelerators in the 60's (see Peter Galison's "Image and Logic").
I worry about the following scenario for Cyrface: a capable programmer hooks up the latest and greatest Bioconductor package to Cytoscape, exposes as best they can the parameterizations offered by that package, and turns the tool over to their collaborating biologist. Experimental data is loaded and analyses or simulations run. Puzzles and inconclusive results will inevitably emerge, requiring detailed knowledge of both the strengths and weaknesses of the Bioc package. With good luck, perseverance and good data, this small working team may in time settle on a satisfactory Cyrface tool which can be reused without the constant intervention of the programmer. This will last until new data is acquired, upsetting the equilibrium, and the hybrid style of work and the back-and-forth between biologist and programmer, begins anew.
I say that I worry about this scenario. It may be exactly the intended use of Cyrface; the problem it is intended to solve. But this essentially sociological characteristic (requirement?) of Cyrface is not described in the paper. I think that those of us who create bioinformatics software tend to avoid being explicit about this - and I think that this (the social & collaborative requirements of bioinformatic research) deserves a lot more attention.
If indeed network biology, as I claim, is in its infancy, then it may be helpful if the ecosystem of Cytoscape-related tools are considered from this perspective. I suspect that the conclusions we might all (mostly) agree upon are:
User-friendly exploration of data-rich networks in a web browser (as with cytoscape.js) will become increasingly popular.
That user-friendliness often competes with analytical nuance and close scrutiny - biologically and clinically useful results become less likely.
Cyrface's connection of Cytoscape to R is a great step in the right direction, marrying as it does user-friendliness with some new analytical power in a way that is nicely complementary to Cytoscape java plugins and Cytoscape access to web services.
Thus, Cyrface is a good step in the right direction. The next steps, it seems to me, include:
Providing easy connections to R (python, C++) analyses for cytoscape.js
A standard mechanism whereby scripts (R, python, Ruby, Perl) upon execution, can start up a Cytoscape or cytoscape.js session, customize it with networks, functions, buttons and menus, and with both public and laboratory data. As a generalization of Cyrface, this mechanism would encourage the rapid expansion of Cytoscape capabilities.
These possible next steps carry on in the spirit of Cyrface, RCytoscape, and Cytoscape3 apps, and will promote the creation of, and sharing of, custom network analyses, shared tools, and lead to fruitful collaborations across the hybrid community of biologists, physicians and programmers.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Shannon PT. Reviewer Report For: Cyrface: An interface from Cytoscape to R that provides a user interface to R packages [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2013, 2:192 (https://doi.org/10.5256/f1000research.2371.r1863)
Bidaut G. Reviewer Report For: Cyrface: An interface from Cytoscape to R that provides a user interface to R packages [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2013, 2:192 (https://doi.org/10.5256/f1000research.2371.r2157)
The paper is well written, however, many items require clarification:
My main criticism is the initial need to install several packages (RCaller, Rserve, Runiversal, Java, etc...), plus Cytoscape and the CommandTool plugin. Since I did not know if this would break
... Continue reading
The paper is well written, however, many items require clarification:
My main criticism is the initial need to install several packages (RCaller, Rserve, Runiversal, Java, etc...), plus Cytoscape and the CommandTool plugin. Since I did not know if this would break my personal configuration or if I would be able to uninstall it, I was not able to performe the whole installation myself. It would be great if the authors could provide a virtual machine image with all the software preloaded so that one can try it out of the box without installation. I know by experience that cityscape plugins tend to work only with a single version of Cytoscape, so a clear list of all the required versions in the paper itself would be very useful.
The number of packages we are dealing with is very confusing. I am also afraid that with such a large number of dependencies, the program may break after any update.
In the Implementation section, the authors mention the "iris dataset". It would be useful to define what this is. It is also mentioned in the documentation but it is still unclear what the authors are referring to. The documentation shows an example where the ggplot2 package is used to plot "petal". Could the authors please define what this is?
Figure 2: It would be useful to define more accurately what type of data we are looking at here (e.g. gene expression?).
In the discussion, several R packages are mentioned (DvD, Cytocopter), what are their links with the present software other than the fact that they run under R? Is it really possible to interact with them from Cytoscape? If so, the proper documentation or a tutorial should be provided. It would also be useful if an actual example using DvD along interaction data, could be shown.
I do not understand the example with DataRail. The usefulness of the example given is not clear to me since it seems that no interaction data is given (for me this is the main purpose of Cytoscape). A useful example for most users might be:
- A network of protein-protein interaction (PPI) data in Cytoscape. - Some expression data in R (an exprs object for instance).
An example of a question might be "How to superimpose the expression data and generate a proper network attribute from it?"
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however I have significant reservations, as outlined above.
Bidaut G. Reviewer Report For: Cyrface: An interface from Cytoscape to R that provides a user interface to R packages [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2013, 2:192 (https://doi.org/10.5256/f1000research.2371.r2157)
Schultz N and Aksoy BA. Reviewer Report For: Cyrface: An interface from Cytoscape to R that provides a user interface to R packages [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2013, 2:192 (https://doi.org/10.5256/f1000research.2371.r1866)
In this manuscript, the authors describe the Cytoscape plugin Cyrface. Cyrface consists of two components: 1) a Java API, which is already being used by the Cytoscape plugins CytoCopteR and DrugvsDisease (both developed by the same group), and 2) a
... Continue reading
In this manuscript, the authors describe the Cytoscape plugin Cyrface. Cyrface consists of two components: 1) a Java API, which is already being used by the Cytoscape plugins CytoCopteR and DrugvsDisease (both developed by the same group), and 2) a graphical user interface that connects R to Cytoscape. As a proof-of-concept to what kind of applications can be built on top this interface, the plugin also supports the MIDAS and SBML-Qual formats.
The article is well written and the tool is useful to the community. However, we recommend the following changes to the article to make it more appealing to potential future users:
As the new version of the Cytoscape (3.x) is becoming more widely used by the community, the authors should explicitly state that they are targeting version 2.8 with this framework. This will reduce the confusion for users who are not as familiar with Cytoscape.
The tutorial in the supplementary materials helps to understand the general use case for this plug-in, but the lack of downloadable “sample” files for this example will make it harder for users to learn how to use the DataRail pipeline. We think it is important to provide example files that people can use for reproducing the figures in the manuscript.
The Cyrface interaction layer with R looks helpful for programmers, but can the authors comment on how these classes are different from the default Java implementations of the RServe clients, e.g. http://rforge.net/RServe? This will help clarify why people should use Cyrface for their next project.
The command line interface (commandTool) appears to be useful; but it seems that it is only capable of running commands in an isolated environment, with each command having its own session. If this is the case, can the authors comment on what the advantage of running R commands from the commandTool is compared to initiating a terminal window and running commands directly from an R shell? Are users able to, for example, pass node/edge attribute fields to the corresponding R commands?
It looks like the current implementation does not support setting a different RServe location other than localhost. Although not necessary, if users are given the option to set a different RServe address within the plug-in, this will further lower the barrier for users who are not experienced with R to use Cyrface, where they can use a pre-installed Rserve hosted on a different machine.
Competing Interests: No competing interests were disclosed.
We confirm that we have read this submission and believe that we have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Schultz N and Aksoy BA. Reviewer Report For: Cyrface: An interface from Cytoscape to R that provides a user interface to R packages [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2013, 2:192 (https://doi.org/10.5256/f1000research.2371.r1866)
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles


Comments on this article Comments (0)