Keywords
GFP, gene function prediction, machine translation
GFP, gene function prediction, machine translation
Determining the functions of proteins is a central problem in biology. There are many databases like RefSeq (http://www.ncbi.nlm.nih.gov/refseq/) that store amino acid sequences and their corresponding functions. However, almost all protein functional prediction methods rely on the identification, characterization, or quantification of sequence similarities between the available proteins of interest1. Even sequences that are similar do not necessarily have identical function. A sequence may be similar to many other sequences, so it can be difficult to choose the most appropriate one. In addition, there is no way to deduce function if there are no similar sequences in any available database.
At present, many machine learning research methods are being applied to deal with the above mentioned problems. Two examples are, Support Vector Machines (SVM); a supervised learning model which is mainly used for classification and regression analysis, and network-based methods such as protein interactome networks2,3. In addition, the sequence features, structures, evolution of amino acid sequences, and other characteristics are also utilized for functional prediction4,5.
The same sequence may have different meanings in different contexts. Different sequences may also represent the same meaning. These problems are normally called “lexically/structurally ambiguous” in the language translation field. Current machine translation research could effectively avoid these problems by building a translation model between two ‘languages’.
Since many amino acid sequences and their corresponding functional description are already publically available, it is possible to build their translation relationships by mature machine translation technology. We address this task in this paper. When presented with a new amino acid sequence we aimed to provide a function-based translation.
To build the model, we need to construct the “parallel corpus” of amino acid sequences and their functional descriptions. The term “parallel corpus” is typically used in linguistic areas to refer to texts that are translations of each other. A parallel corpus contains the data for two languages.
For example, a parallel corpus from English to French translation:
English sentence: I love apples
French sentence: J’adore les pommes
Here, we selected amino acid sequences and their functional descriptions in Gene Ontology form to build the corpus6. The reason for using the Gene Ontology is that it gives the unified descriptions for protein functions of all species. The Gene Ontology data can be found on the website www.geneontology.org (http://www.geneontology.org/GO.downloads.annotations.shtml). The corresponding amino acid sequence data can be downloaded from the website www.uniprot.org (http://www.uniprot.org/downloads), which is a central repository of amino acid sequence. We use the database ID to identify a sequence and find its correspondences in above two data sources.
An example of a parallel corpus from “amino acid sequence” to “function” translation:
Amino acid sequence (amino acid):
MTMDKSELVQKAKLAEQAERYDDMAAAMKAVTEQGHELSNEERNLLSVAYKNVV
Function description (Gene ontology term ID):
0005737 0006605 0019904 0035308 0042470 0042826 0043085
The basic idea of machine translation is very simple. It tries to infer “rules” from the parallel corpus and build a translation model. The “rules” are mainly the words or phrase correspondences between two languages. Similar sequences will get near translation results. Although the application of translation technology in protein functional prediction seems reasonably simple, as far as we know, there is no existing program that applies machine translation to protein sequence function analysis.
Here we build three groups of gene parallel corpora. Group 1 only contains human amino acid data. It has 45,538 amino acid sequences and related Gene Ontology descriptions. Group 2 is a mixed dataset of several model species. It contains about 100,000 amino acid sequences. Group 3 is the largest dataset. It contains more than 2,000,000 amino acid sequences. All of the corpora can be downloaded from www.geneontology.org (Gene Ontology data) and www.uniprot.org (amino acid sequence data). We also provide these Gene ontology and amino acid sequence data in ‘data’ directory of Supplementary material with this paper.
First, we need to “clean” the corpus. Data cleaning is the process of detecting and correcting (or removing) corrupted or inaccurate records from a dataset. For machine translation, sequences with significant differences in length between the sources and targets should be removed. The empty and overlong sequences should also be deleted. For amino acid data, we deleted long sequences (amino acid sequence length<1000 (amino acids), Gene ontology terms number<100) and empty sequences.
After data cleaning, we had about 14,000 pieces of data for 45,538 human amino acids. Data of groups 2 and 3 are also reduced to about 1/4 of the original data size.
Amino acid sequences are not naturally segmented. So we still need to “segment” them.
Text segmentation means dividing a string of written language into its component words. In English and many other languages using some form of the Latin alphabet, the space is a good approximation of a word delimiter. But for some East-Asia languages like Chinese, there are no natural delimiters. To process these languages, we need to segment sentences into “words” sequences first. For example:
A Chinese sentence:
我愛苹果(Iloveapple)
We need its word sequence form:
我 愛 苹果(I love apple)
Segmenting methods on processing East-Asia languages are applied into amino acid sequence segmentation. Here we segment/tokenise amino acid sequences by an unsupervised segmentation method9. Its main idea is to evaluate the probabilities of all possible words by Expectation Maximization (EM), an iterative method for finding maximum likelihood estimates of parameters in statistical models10, or other statistical methods like the n-grams language method11. Then for a sequence, we select the segmenting form with maximum probability as the segmentation of this sequence.
An example of parallel corpus about “segmented amino acid sequence” to “function”:
Amino acid sequence (amino acid, segmented):
MTMDKS ELVQKA KLAEQA ERYDDM AAA MKAVTE QGH ELSNEE RNLLSV AYKNVV
Function description (Gene ontology terms ID):
0005737 0006605 0019904 0035308 0042470 0042826 0043085
After segmenting, we also delete the obviously mis-aligned sequences (‘word’ number ratio>9. For above example, there are 10 amino acid words and 7 Gene ontology word. So its word number ratio is 10/7) in corpus.
There have been many open source translation systems. Normally, the Moses system is treated as the baseline system12. Moses is a statistical machine translation system that allows you to automatically train translation models for any language pair. All you need is a collection of translated texts (parallel corpus). Once you have a trained model, an efficient search algorithm quickly finds the highest probability translation among the exponential number of choices. Here we also use the Moses system to build the translation model for amino acid sequence to Gene ontology terms. Moses offers two types of translation models: phrase-based and tree-based. We selected the phrase-based model.
We used 95% of the data to train the model, and the remaining 5% of the data for testing.
The input for Moses is the parallel corpus. All parameters are set by default values (more details reference in the “data” and “code” sections of the Supplementary materials). The language model for the Gene ontology sequence is set as 2-grams (in the fields of computational linguistics and probability, an n-gram is a contiguous sequence of n items from a given sequence of text. For more information, see Xiaping Ge, Wanda Prat, and Padhratic Smyth10). Then we could get an amino acid sequence to Gene ontology translation model. The detailed process is provided in the ‘code’ directory of Supplementary materials. Its main steps are described as in Figure 1.
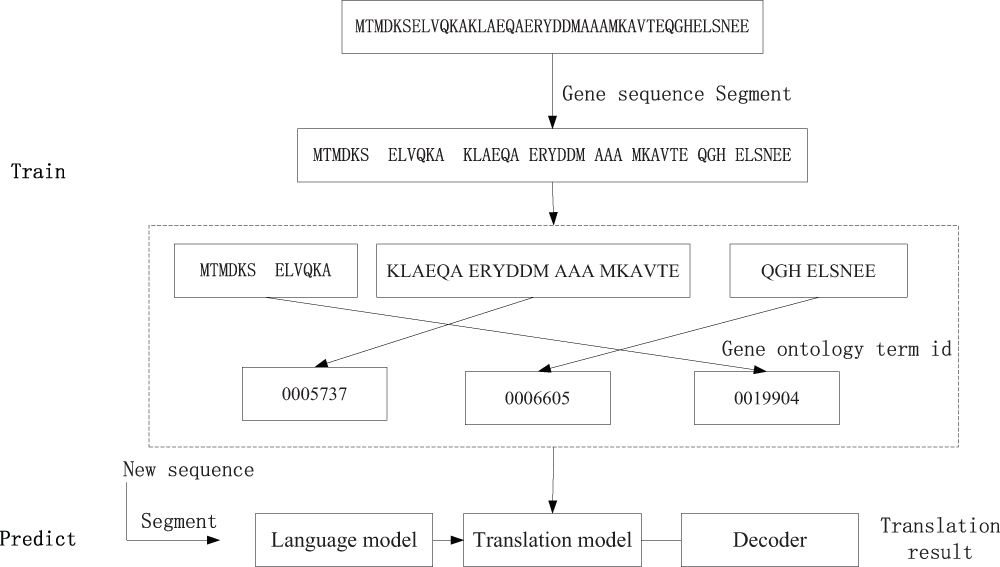
We used the Bilingual Evaluation Understudy (BLEU) score to judge the performance of the translation system13. BLEU is an algorithm for evaluating the quality of text which has been machine-translated from one natural language to another. The main idea of BLEU score can be described by formula 1.
In this formula m is the number of words from the candidate translation that are found in the reference translation, and wt is the total number of words in the candidate translation.
For example, a candidate translation is “I air apple”. The reference translation is “I love apple”. There are 3 words in the candidate translation. Two words, “I” and “apple”, appear in the reference translation. So the BLEU score of this translation is 2/3=0.67.
The improved version of BLEU normally considers the n-grams match. Since we don’t care about the order of Gene ontology words, we only needed to use the unigram form of BLEU.
Because different amino acid sequence segmentation methods will produce completely different “word” sequences, and the segment methods mainly rely on the selection of maximal word length, we tried different maximal word lengths. The relation of maximal word length and the BLEU score are shown in Figure 2.
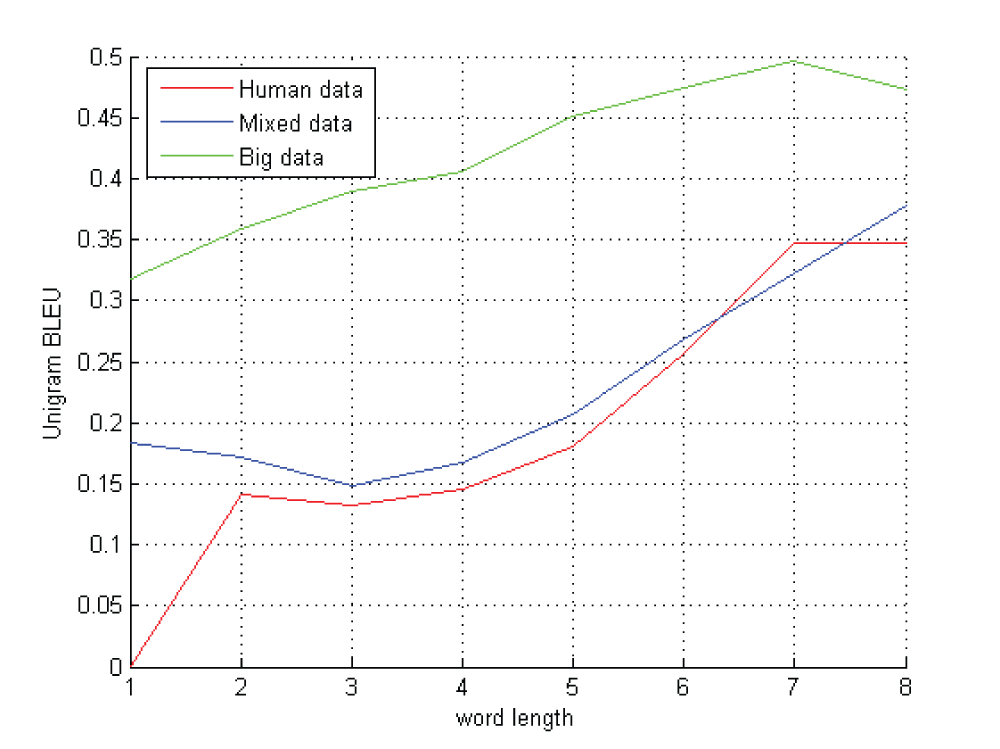
The red line corresponds to group 1, human dataset. Blue line is group 2, mixed dataset and green is group 3, the big dataset.
Figure 2 shows maximal word length of 7 or 8 letters is a good choice for amino acid translation. In theory, we need 20n letters corpus to train an n word length segmenting model. For n=8, we need about 26 Gigabytes amino acid sequence data. For n=9, we need 512 Gigabytes of data. Now there are only about 20 Gigabytes in annotated amino acid sequence data in Gene ontology databases (http://www.geneontology.org/GO.downloads.annotations.shtml). So a practical word length selection could be 7.
If we don’t segment the amino acid sequence and treat every amino acid letter as a “word”, 20+ amino acid letters could only express 20+ functions. But from Figure 2, we deduct that we could still get a translation model if we have sufficient corpus. This was mainly because the training system could combine some consecutive letters into “phrases” in the alignment process. So such “phrases” could represent more things.
In Figure 2, we could also find the increasing of the size of corpus could obviously improve the performance of translation. So far now, we tried a 5,000,000 amino acid sequences data set. Its BLEU score could reach 0.6. We also provide the method to build 40,000,000 amino acid sequences corpus data in ‘get_gene_data’ directory of supplied materials.
For a usable translation system, its unigram BLEU score should be more than 0.5. Our amino acid sequence to Gene ontology translation model has reached this level. So we could compare it with conventional search based method.
The sections above show that the “amino acid sequence” to “protein function” translation is workable. Next, we compared it with current protein functional prediction methods.
We designed the comparison as follows:
(1) Dataset: we divided the parallel corpus into 95% training corpus and 5% test corpus.
(2) For the translation-based method: we trained a “amino acid sequence” to “Gene ontology” translation model based on the training corpus and then predicted the functions of a test amino acid sequence based on this translation model.
(3) For the search-based method: we indexed the amino acid sequences in the training corpus by BLAST to build a training sequences database. To predict the function of the test sequences, we searched the test amino acid sequence in database of training sequences respectively. The Gene ontology description of the best match sequence was regarded as the function of this test sequence.
(4) Comparing method: we compared the predicted functions of two methods by the BLEU score. The results are shown in Table 1.
| Human data (group 1) | Mixed data (group 2) | Big data (group 3) | |
|---|---|---|---|
| Search method | 0.41 | 0.54 | 0.83 |
| Translation method | 0.35 | 0.37 | 0.50 |
From Table 1, we found that the functional prediction performance of the translation method was still not as good as BLAST, but there were about 1%–5% sequences whose functions the BLAST method could not predict. On the other hand, the translation method could predict the functions of all amino acid sequences. Here, we mainly want to show the feasibility of the new method. More datasets and translation methods should be tested in the future.
Based on statistical machine translation technology, we present a novel method to predict the function of amino acid sequences. Although its performance remains to be improved, it shows that the application of machine translation in protein functional prediction promising. For statistical translation methods, the more corpuses we generate, the better results we would obtain. Google’s translation system has proven that vast amounts of corpus with a few rules, or even without any rules, can produce excellent translation results14. Since the protein functional prediction problem is so close to ‘language translation’, translation based protein functional prediction is likely to be the most promising approach. Source codes are permanently accessible from 10.5281/zenodo.7506.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)