Gautam S and Gupta MN. Solid state fluorescence of proteins in high throughput mode and its applications [version 1; peer review: 2 approved with reservations]. F1000Research 2013, 2:82 (https://doi.org/10.12688/f1000research.2-82.v1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
1Department of Chemistry, Indian Institute of Technology Delhi, New Delhi, 110016, India
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
A
simple method to determine fluorescence emission spectra of proteins in solid
state is described. The available commercial accessories can only accommodate
solid samples and hence do not allow a direct comparison between fluorescence
spectra of a sample in solution and solid state form. Such comparisons are
valuable to monitor the changes in protein structure when it is “dried” or immobilized on a solid surface (for biocatalysis or sensor applications). The
commercially available accessories also do not allow working in a high
throughput mode. We describe here a simple method for recording fluorescence
emission spectra of protein powders without using any dedicated accessory for
solid samples. This method works with a 96-well plate format. It enables the
comparison of fluorescence spectra of a sample in a solid state with solution
spectra, using comparable quantities of protein. The fluorescence emission
spectra were blue-shifted (4 to 9 nm), showed an increase in the intensity for different
proteins studied upon lyophilization, and were similar to what has been reported
by others using available commercial accessories for solid state samples. After
validating that our method worked just as well as the dedicated accessories, we
applied the method to compare the fluorescence emission spectra of α-chymotrypsin
in solution, precipitated form and the lyophilized powder form. α-Chymotrypsin
in solution showed a λmax of 335 nm while a high-activity
preparation of the same enzyme for non-aqueous media, known as enzyme
precipitated and rinsed with propanol (EPRP), showed an increase in the
intensity of the fluorescence emission spectra. However, there was a small red
shift of 2 nm (λmax of 337 nm) in contrast to lyophilized powder
which showed a λmax of 328 nm. This is due to a difference in the
tertiary structure of the protein as well as the microenvironment of aromatic
residues between the two preparations. We further examined the fluorescence
emission spectra of green fluorescent protein (GFP) in solution and solid form.
The relative fluorescence intensity of lyophilized GFP powder was decreased significantly
to 17% as compared to GFP in solution, and showed a red shift of 4 nm in the emission
λmax. It was found that fluorescence resonance energy transfer (FRET)
between tryptophan (Trp57) and the cyclic chromophore of GFP was significantly
diminished. This indicated the change in the microenvironment around the cyclic
chromophore in GFP upon lyophilization.
Corresponding author:
Munishwar N Gupta
Competing interests:
No competing interests were disclosed.
Grant information:
This work was funded by research support from the Department of Biotechnology (DBT) [Grant number: BT/PR14103/BRB/10/808/2010] and the Department of Science and Technology (DST) [Grant number: SERB/F/1776/2011-2012], Government of India. Financial support was also provided by the Council of Scientific and Industrial Research to SG in the form of a Junior Research Fellowship.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Fluorescence spectroscopy is a powerful tool to study protein structure1–3. Measurement of the fluorescence of proteins, when the latter is present in the solid state, is useful in several different contexts. Solid state fluorescence has a number of uses including in protein assays with protein electrophoresis samples4, enzyme immobilization5, microscopy6, detecting changes in protein tertiary structure upon lyophilization7, sensors and microarrays8,9 and characterizing solid waste10. Of these, fluorescence measurement of lyophilized samples is itself valuable for a variety of different kinds of studies. Enzyme catalysis in low water media is often carried out with lyophilized enzyme powders11–14. Only recently, circular dichroism (CD) of α-chymotrypsin “dried” (bulk water removed) with different methods has been reported with the help of a special accessory for recording CD spectra of solid samples as suspensions15.
Some commercially available accessories for spectrofluorimeters allow recording the fluorescence emission spectra of the solid samples7,16–19. These available commercial accessories can only accommodate solid samples and hence do not allow a direct comparison between fluorescence spectra of a sample in solution and solid state form. These accessories also do not allow working in a high throughput mode.
We describe here a simple method for recording fluorescence emission spectra of protein powders without using any dedicated accessory. This method works with a 96-well plate format. It enables the comparison of fluorescence spectra of a sample in a solid state with solution spectra, using comparable quantities of protein. It was found that, just like spectra recorded with these commercial accessories, the spectra of lyophilized powders obtained by our method showed a blue shift of λmax (as compared to the solution spectra). After this validation, the method was used for two specific applications. In the first case, the method was used for assessing the tertiary structure of “dried” α-chymotrypsin. It was also used to track changes in fluorescence spectra of green fluorescent protein (GFP) when it is dried. While the former application is relevant to non-aqueous enzymology, the latter provides some insight into fluorescence resonance energy transfer (FRET) between tryptophan of GFP (Trp57) and its cyclic chromophore20,21.
These illustrative examples show that the valuable information provided by fluorescence emission spectroscopy about conformational changes in proteins upon drying can be obtained in a simple manner by anybody with a fluorescence-based microplate reader.
Materials
Ampicillin, bovine serum albumin (BSA, cat. no. A7030), α-chymotrypsin (protease from bovine pancreas, cat. no. C4129), lysozyme (from chicken egg white, cat. No. L6876), phenylmethanesulfonylfluoride (PMSF) and n-propanol were purchased from Sigma-Aldrich (St. Louis, MO, USA). Isopropyl β-D-thiogalactopyranoside (IPTG) and LB medium were obtained from Himedia Laboratories (Mumbai, India). TLL (Thermomyces lanuginosus lipase) was a kind gift from Novozymes (Denmark). Candida rugosa lipase was a gift from Amano Enzyme Inc. (Nagoya, Japan). Ninety-six well polystyrene microplates were obtained from Porvair Sciences (Leatherhead, UK). All other chemicals used were of analytical grade. All the proteins used were >95% pure on SDS-PAGE.
Overexpression and isolation of GFP
The plasmid pGFPuv expressing recombinant GFP was transformed into E. coli BL21 (DE3)22. A single colony was picked and inoculated into 5 mL LB medium containing 100 μg mL-1 ampicillin. In total, 1% of primary inoculum was transferred into 1 L fresh LB broth (amp+) and grown at 37°C with shaking at 200 rpm until absorbance at 600 nm reached 0.8. Induction was carried out by adding 1 mM isopropyl β-D-thiogalactopyranoside (IPTG) (final concentration). The culture was further grown under similar conditions for 12 h. The cells were harvested by centrifugation at 8000g for 10 min at 4°C. GFP was isolated from E. coli cells by sonication in 50 mM phosphate buffer, pH 7.5, containing 2 M NaCl and 100 μM phenylmethanesulfonylfluoride (PMSF), three times with 15 s pulses on ice, and centrifugation at 9000g for 10 min at 4°C. The supernatant thus obtained was used as a crude extract for GFP and further purified to homogeneity (as shown by single band on SDS-PAGE) by immobilized metal affinity chromatography using nickel-alginate beads as described earlier22.
Lyophilization
Lyophilization of all the proteins was carried out on a freeze dryer from Allied Frost (New Delhi, India). Proteins were dialyzed against buffer (10 mM Tris-HCl, pH 7.0 for BSA, TLL, lysozyme, CRL and α-chymotrypsin; and 10 mM phosphate buffer, pH 7.5 for GFP) and were frozen at -70°C for 1 h before lyophilization.
Preparation of enzyme precipitated and rinsed with propanol (EPRP) of α-chymotrypsin
Enzyme precipitated and rinsed with propanol (EPRP) of α-chymotrypsin was prepared as described previously15. A total of 4 mg of α-chymotrypsin was dissolved in 400 µL of 10 mM Tris-HCl buffer, pH 7.8. Enzyme solution was then added drop wise to 4 ml of n-propanol with stirring at 4°C. After addition, the suspension was stirred for 30 min at 4°C. The suspension was then centrifuged at 5000g for 10 min at 4°C, and the precipitate was rinsed three times with dry and chilled n-propanol.
Fluorescence measurements
All fluorescence spectra were recorded on a Cary Eclipse, Varian spectrofluorimeter (Varian Inc., Mulgrave, Victoria, Australia) at 25°C by using the microtitre plate reader accessory for reading 96-well microplates. The typical protein concentration of proteins used for fluorescence measurements in solution was 2 mg/mL in a total volume of 200 µL. Proteins were lyophilized at the same concentration and same amount of protein was used for solid state fluorescence measurements. The fluorescence emission spectra were recorded from 300 nm to 400 nm upon excitation at 280 nm2. For GFP, the fluorescence emission spectra were recorded from 450 nm to 600 nm upon excitation at 395 nm23. The excitation and emission slit widths were kept at 2 nm and 5 nm, respectively. All fluorescence spectra were normalized and corrected for background contributions including the buffer.
Estimation of protein concentration
Protein concentration was estimated by the dye binding method using bovine serum albumin as the standard protein24.
Results and discussion
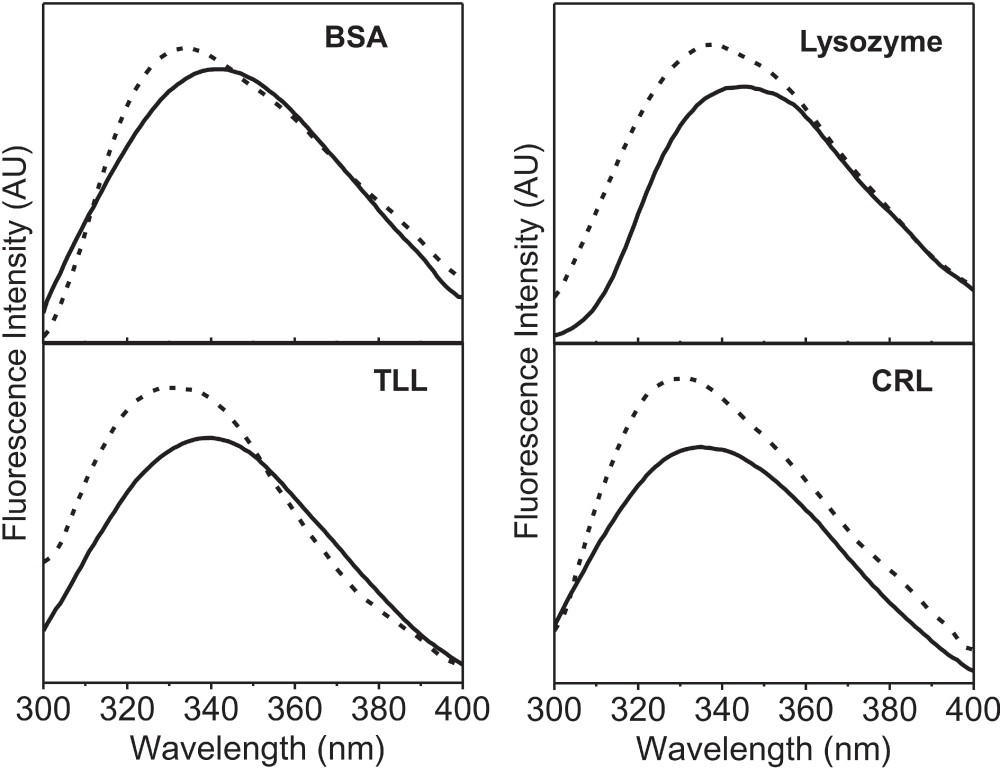
The method developed here consists of simply placing the lyophilized powder of the protein in the well of 96-well microplate. The fluorescence spectra were recorded on a standard Varian microplate reader. The λmax excitation known for the protein solutions were used for solid samples as well. Intrinsic fluorescence emission spectra of four different commercial proteins were obtained after lyophilization from the aqueous buffer and compared with the spectra of the respective protein in the aqueous buffer solution (Figure 1). The amount of protein in each solution was the same as was used for obtaining the lyophilized powders. In all the cases there was a blue shift in emission λmax(Table 1) and an increase in the intensity of the emission spectra of the lyophilized proteins as compared to the protein in aqueous solution. It is important to note that a similar blue shift in the λmax have been reported by Ramachander et al7. while comparing the solid state and solution state fluorescence spectra of four therapeutic proteins (the identities of the proteins were not disclosed by these authors). These workers had used a special accessory (called a solid state holder set up) for the Cary Eclipse spectrofluorimeter. The blue shift in the lyophilized state reflects that the environment of intrinsic fluorophores is more non-polar. This is expected due to the removal of water. The small differences in the extent of the blue shift (Table 1) in case of the four proteins presumably originate from the differences in the microenvironments of tryptophan in the folded structure of each of the proteins. To start with, when in solution, the microenvironments of tryptophan are expected to be different between different proteins.
Figure 1. Fluorescence emission spectra (AU, arbitrary units) of four different proteins [Bovine serum albumin (BSA), Thermomyces lanuginosus lipase (TLL), Candida rugosa lipase (CRL)].
Protein in aqueous buffer (10 mM Tris-HCl, pH 7.0) (—) and lyophilized protein powder (- -). All these fluorescence emission spectra were recorded with excitation at 280 nm using excitation and emission slit widths of 2 nm and 5 nm, respectively.
Table 1. Fluorescence emission λmax (nm) of four different lyophilized proteins and their comparison with the proteins in 10 mM Tris-HCl, pH 7.0.
Protein
λmax (nm) of the protein in aqueous buffer solution
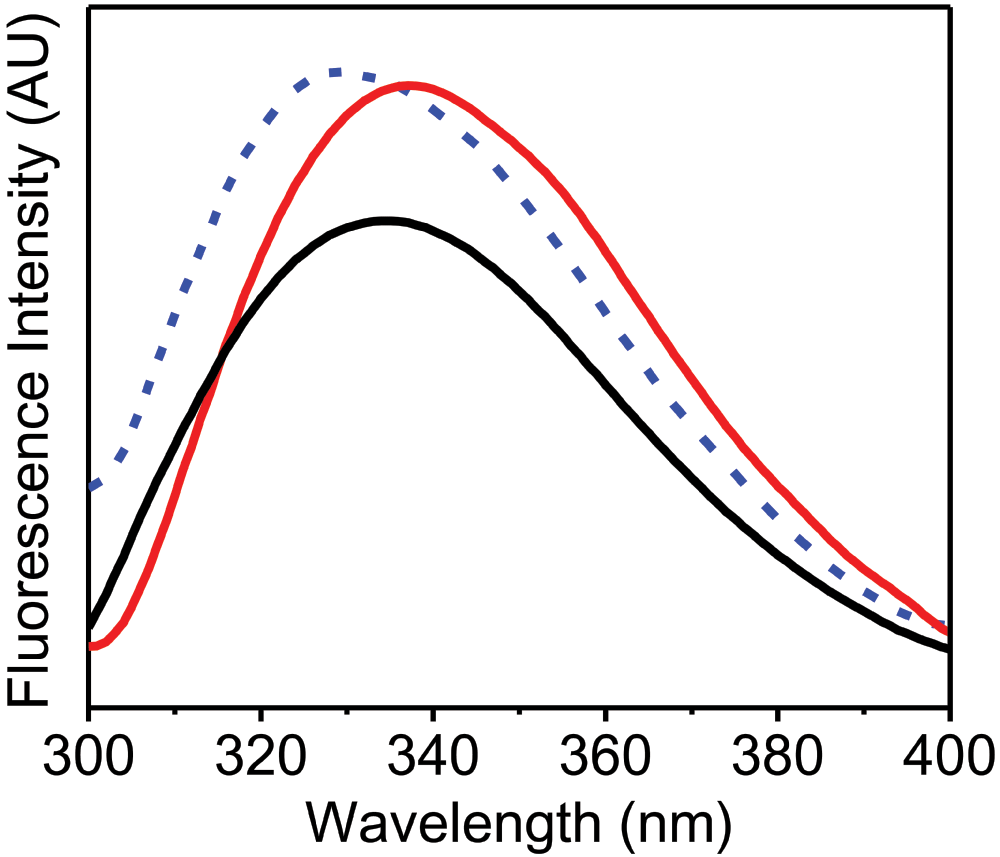
Figure 2 shows the fluorescence emission spectra of α-chymotrypsin in solution and in the solid state. Native α-chymotrypsin in aqueous buffer showed emission λmax of 335 nm while upon lyophilization it was blue shifted to 328 nm with an increase in the intensity. This is likely again due to the non-polar environment of the aromatic residues. It has been shown that lyophilized preparations of α-chymotrypsin show poor esterification/transesterification activity in low water media containing organic solvents25. Low activities of lyophilized powders in such media have been explained due to structural changes which occur upon lyophilization14. “Dry” preparations obtained by precipitation of α-chymotrypsin from its aqueous solution by addition of water miscible organic solvents are known to show much better activities in low water media15,26,27. Recently, Solanki et al15. found that changes in the CD spectra upon “drying” correlated well with catalytic activities in low water media for various α-chymotrypsin preparations. A high activity preparation of α-chymotrypsin for low water media (EPRP)15 showed a very small red shift in the emission λmax to 337 nm with an increase in the intensity of the emission spectra, in contrast to the lyophilized protein which showed a blue shift. This further highlights that the lyophilized protein is different from the high activity preparation (EPRP) in terms of the tertiary structure, demonstrating that the simple fluorescence method proposed here can successfully monitor changes in the tertiary structure of different types of formulations of solid proteins.
Figure 2. Fluorescence emission spectra (AU, arbitrary units) of α-chymotrypsin.
α-Chymotrypsin in aqueous buffer (10 mM Tris-HCl, pH 7.0) (black curve), lyophilized α-chymotrypsin powder (blue dashed curve) and solid sample of enzyme precipitated and rinsed with propanol (EPRP) of α-chymotrypsin (red curve). All these fluorescence emission spectra were recorded with excitation at 280 nm using excitation and emission slit widths of 2 nm and 5 nm, respectively.
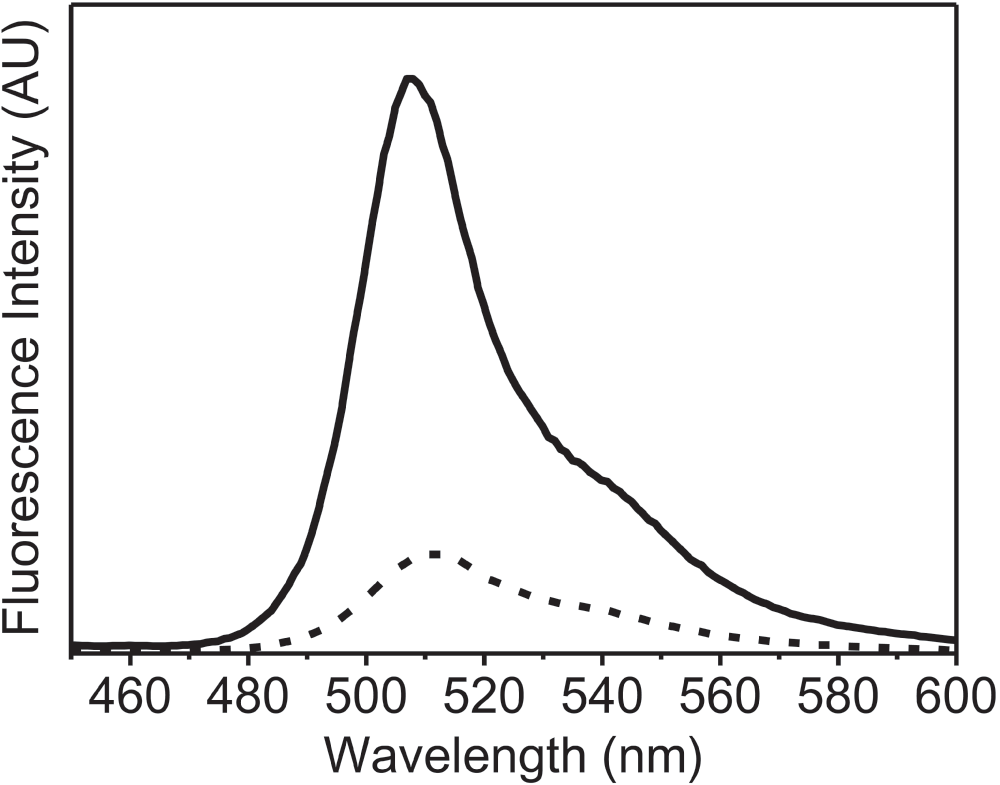
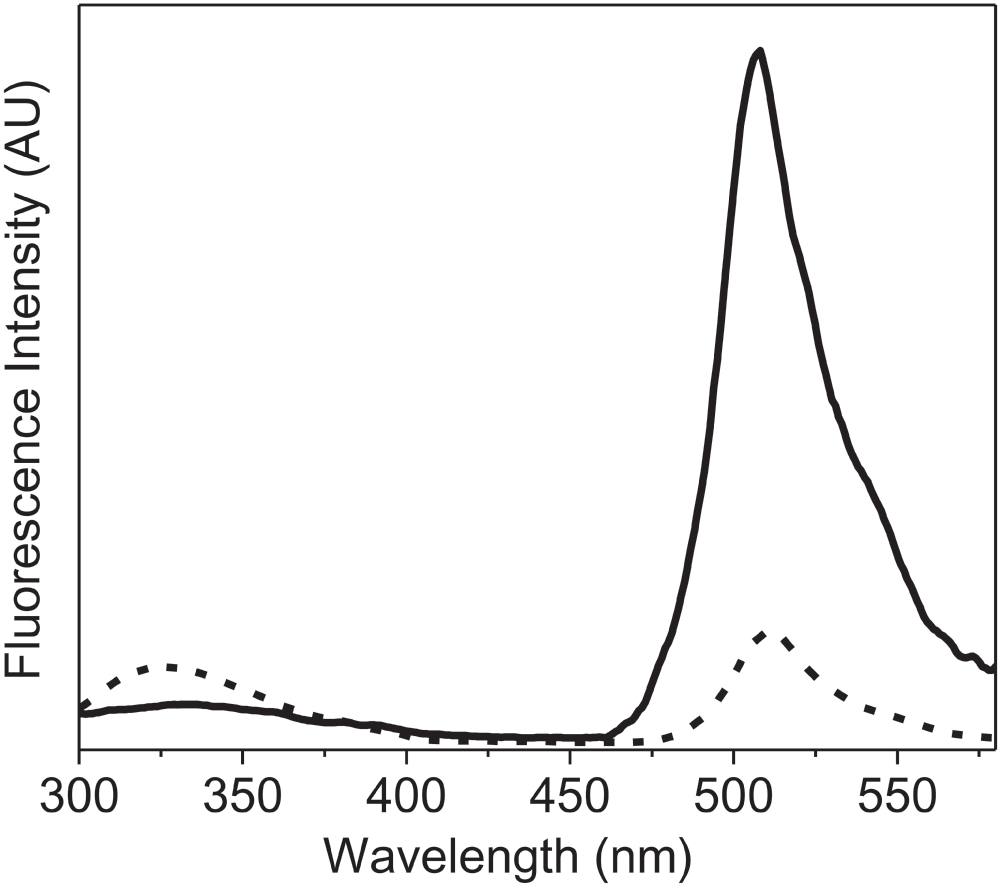
To further examine the application of this new method, we recorded the fluorescence spectra of the lyophilized formulation of recombinant GFP. In this case, the high intrinsic fluorescence of GFP due to the cyclic moiety present in the protein, which is very sensitive to changes in the structure of protein23, was studied (Figure 3). The lyophilized protein showed a considerable decrease in the intensity of the fluorescence emission spectra to 17% as compared to that of GFP in solution. The emission λmax was also slightly red shifted upon lyophilization (4 nm, from 508 nm to 512 nm). These changes (in fluorescence intensity and shift of λmax emission) were opposite to what was observed with other proteins (Figure 1). Visser et al20. have shown that the fluorescence of the cyclic chromophore in GFP results from the energy transfer from the tryptophan. Figure 4 shows that the energy transfer between the tryptophan and the cyclic chromophore is much less in the lyophilized form. It is noteworthy that the change in the emission intensity due to tryptophan residues (at ~340 nm) was observed in GFP (Figure 4), just as for the other proteins (Figure 1). Both changes reflect how the microenvironment affects the emission fluorescence of the unique chromophore of GFP and could be due to the degradation of this cyclic chromophore upon lyophilization.
Figure 3. Fluorescence emission spectra (AU, arbitrary units) of green fluorescent protein (GFP).
GFP in aqueous buffer (50 mM PBS) (solid line) and lyophilized powder of GFP (dashed line). These fluorescence emission spectra were recorded with excitation at 395 nm using excitation and emission slit widths of 2 nm and 5 nm, respectively.
Figure 4. Fluorescence emission spectra (AU, arbitrary units) of green fluorescent protein (GFP) showing fluorescence resonance energy transfer (FRET) between tryptophan (Trp57) and cyclic chromophore.
GFP in aqueous buffer (50 mM PBS) (solid line) and lyophilized powder of GFP (dashed line). These fluorescence emission spectra were recorded with excitation at 295 nm using excitation and emission slit widths of 2 nm and 5 nm, respectively.
Conclusion
A simple method of placing the dry protein powder in a 96-well microplate enables the generation of fluorescence spectra of a protein in the solid state. As the fluorescence spectra of the solution can also be recorded in an identical fashion, the exact comparison between the solution and solid state spectra is possible.
Author contributions
MNG designed the study. MNG and SG participated in the interpretation of data and the writing of the manuscript. SG carried out the experimental work. Both authors approved the submission of the final manuscript.
Competing interests
No relevant competing interests where disclosed.
Grant information
This work was funded by research support from the Department of Biotechnology (DBT) [Grant number: BT/PR14103/BRB/10/808/2010] and the Department of Science and Technology (DST) [Grant number: SERB/F/1776/2011–2012], Government of India. Financial support was also provided by the Council of Scientific and Industrial Research to SG in the form of a Junior Research Fellowship.
Acknowledgments
The financial support provided by the Council of Scientific and Industrial Research to SG in the form of a Junior Research Fellowship is gratefully acknowledged.
References
1.
Royer CA: In: Shirley BA (ed) Protein stability and folding. Humana Press, Totowa, New Jersey (1995).
This work was funded by research support from the Department of Biotechnology (DBT) [Grant number: BT/PR14103/BRB/10/808/2010] and the Department of Science and Technology (DST) [Grant number: SERB/F/1776/2011-2012], Government of India. Financial support was also provided by the Council of Scientific and Industrial Research to SG in the form of a Junior Research Fellowship.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Gautam S and Gupta MN. Solid state fluorescence of proteins in high throughput mode and its applications [version 1; peer review: 2 approved with reservations]. F1000Research 2013, 2:82 (https://doi.org/10.12688/f1000research.2-82.v1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Uversky V. Reviewer Report For: Solid state fluorescence of proteins in high throughput mode and its applications [version 1; peer review: 2 approved with reservations]. F1000Research 2013, 2:82 (https://doi.org/10.5256/f1000research.872.r838)
The suggested technique for the analysis of solid state fluorescence of proteins is simple and can find multiple applications. Although the authors analyzed both fluorescence intensity and λmax of protein powders and corresponding protein solutions, the applicability of fluorescence intensity
... Continue reading
The suggested technique for the analysis of solid state fluorescence of proteins is simple and can find multiple applications. Although the authors analyzed both fluorescence intensity and λmax of protein powders and corresponding protein solutions, the applicability of fluorescence intensity is questionable. In fact, fluorescence intensity depends on a wide range of factors and cannot be easily interpreted, especially if samples are in different aggregated states (solid versus solution). Therefore, only λmax should be taken as a parameter for analysis since this characteristic has understandable physical grounds.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however I have significant reservations, as outlined above.
Uversky V. Reviewer Report For: Solid state fluorescence of proteins in high throughput mode and its applications [version 1; peer review: 2 approved with reservations]. F1000Research 2013, 2:82 (https://doi.org/10.5256/f1000research.872.r838)
Munishwar N Gupta, Department of Chemistry, Indian Institute of Technology Delhi, New Delhi, 110016, India
15 Mar 2013
Author Response
The
references provided in the manuscript to the earlier work carried out with
commercial accessories described differences between the solution spectra and
the solid state spectra and list changes in
...
Continue readingThe
references provided in the manuscript to the earlier work carried out with
commercial accessories described differences between the solution spectra and
the solid state spectra and list changes in both intensity and λmax
shift. In order to validate our method, we wanted to point out that
both/similar changes occur with our method as well. However, as Table 1 of our
manuscript shows, we have focused on the peak position. The amended manuscript
can point out that intensities can be affected by many variables and one should
rely more upon the λmax position.
These explanations
can be added in the amended manuscript.
The
references provided in the manuscript to the earlier work carried out with
commercial accessories described differences between the solution spectra and
the solid state spectra and list changes in both intensity and λmax
shift. In order to validate our method, we wanted to point out that
both/similar changes occur with our method as well. However, as Table 1 of our
manuscript shows, we have focused on the peak position. The amended manuscript
can point out that intensities can be affected by many variables and one should
rely more upon the λmax position.
These explanations
can be added in the amended manuscript.
Competing Interests:No competing interests were disclosed.Close
Munishwar N Gupta, Department of Chemistry, Indian Institute of Technology Delhi, New Delhi, 110016, India
15 Mar 2013
Author Response
The
references provided in the manuscript to the earlier work carried out with
commercial accessories described differences between the solution spectra and
the solid state spectra and list changes in
...
Continue readingThe
references provided in the manuscript to the earlier work carried out with
commercial accessories described differences between the solution spectra and
the solid state spectra and list changes in both intensity and λmax
shift. In order to validate our method, we wanted to point out that
both/similar changes occur with our method as well. However, as Table 1 of our
manuscript shows, we have focused on the peak position. The amended manuscript
can point out that intensities can be affected by many variables and one should
rely more upon the λmax position.
These explanations
can be added in the amended manuscript.
The
references provided in the manuscript to the earlier work carried out with
commercial accessories described differences between the solution spectra and
the solid state spectra and list changes in both intensity and λmax
shift. In order to validate our method, we wanted to point out that
both/similar changes occur with our method as well. However, as Table 1 of our
manuscript shows, we have focused on the peak position. The amended manuscript
can point out that intensities can be affected by many variables and one should
rely more upon the λmax position.
These explanations
can be added in the amended manuscript.
Competing Interests:No competing interests were disclosed.Close
Rotello V. Reviewer Report For: Solid state fluorescence of proteins in high throughput mode and its applications [version 1; peer review: 2 approved with reservations]. F1000Research 2013, 2:82 (https://doi.org/10.5256/f1000research.872.r834)
The use of solid state fluorescence to provide protein quality control is a modest technical advance. This study looks at both fluorescence peak position and intensity. The latter value, however, will be affected by light scattering that is both particle
... Continue reading
The use of solid state fluorescence to provide protein quality control is a modest technical advance. This study looks at both fluorescence peak position and intensity. The latter value, however, will be affected by light scattering that is both particle size and morphology dependent. I would suggest focusing on the peak position as this will provide a much better metric.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however I have significant reservations, as outlined above.
Rotello V. Reviewer Report For: Solid state fluorescence of proteins in high throughput mode and its applications [version 1; peer review: 2 approved with reservations]. F1000Research 2013, 2:82 (https://doi.org/10.5256/f1000research.872.r834)
Munishwar N Gupta, Department of Chemistry, Indian Institute of Technology Delhi, New Delhi, 110016, India
15 Mar 2013
Author Response
1. The work is not aimed at suggesting that fluorescence emission spectroscopy should be applied to solid samples of proteins instead of using their solutions. On the other hand, as
...
Continue reading1. The work is not aimed at suggesting that fluorescence emission spectroscopy should be applied to solid samples of proteins instead of using their solutions. On the other hand, as discussed in the manuscript, there are many investigations (enzyme immobilization on solid surfaces, enzyme powders in low water enzymology) wherein structural characterization would be valuable with the solid state samples. The availability of some commercial accessories fulfills this need. So, we offer a way of measuring fluorescence spectra in a 96-well format without any additional accessories.
2. The 96-well plates used were black from all sides except from the top. So, the emission takes place along the same path direction as the exciting radiation. This minimizes scattered light and distortion of the spectra. This results in the spectra which are of reasonable quality as seen in our raw data.
3. The references provided in the manuscript to the earlier work carried out with commercial accessories described differences between the solution spectra and the solid state spectra and list changes in both intensity and λmax shift. In order to validate our method, we wanted to point out that both/similar changes occur with our method as well. However, as Table 1 of our manuscript shows, we have focused on the peak position. In the amended manuscript we will point out that intensities can be affected by many variables and that one should rely more upon the λmax position. These explanations will be added to our amended manuscript.
1. The work is not aimed at suggesting that fluorescence emission spectroscopy should be applied to solid samples of proteins instead of using their solutions. On the other hand, as discussed in the manuscript, there are many investigations (enzyme immobilization on solid surfaces, enzyme powders in low water enzymology) wherein structural characterization would be valuable with the solid state samples. The availability of some commercial accessories fulfills this need. So, we offer a way of measuring fluorescence spectra in a 96-well format without any additional accessories.
2. The 96-well plates used were black from all sides except from the top. So, the emission takes place along the same path direction as the exciting radiation. This minimizes scattered light and distortion of the spectra. This results in the spectra which are of reasonable quality as seen in our raw data.
3. The references provided in the manuscript to the earlier work carried out with commercial accessories described differences between the solution spectra and the solid state spectra and list changes in both intensity and λmax shift. In order to validate our method, we wanted to point out that both/similar changes occur with our method as well. However, as Table 1 of our manuscript shows, we have focused on the peak position. In the amended manuscript we will point out that intensities can be affected by many variables and that one should rely more upon the λmax position. These explanations will be added to our amended manuscript.
Competing Interests:No competing interests were disclosed.Close
Munishwar N Gupta, Department of Chemistry, Indian Institute of Technology Delhi, New Delhi, 110016, India
15 Mar 2013
Author Response
1. The work is not aimed at suggesting that fluorescence emission spectroscopy should be applied to solid samples of proteins instead of using their solutions. On the other hand, as
...
Continue reading1. The work is not aimed at suggesting that fluorescence emission spectroscopy should be applied to solid samples of proteins instead of using their solutions. On the other hand, as discussed in the manuscript, there are many investigations (enzyme immobilization on solid surfaces, enzyme powders in low water enzymology) wherein structural characterization would be valuable with the solid state samples. The availability of some commercial accessories fulfills this need. So, we offer a way of measuring fluorescence spectra in a 96-well format without any additional accessories.
2. The 96-well plates used were black from all sides except from the top. So, the emission takes place along the same path direction as the exciting radiation. This minimizes scattered light and distortion of the spectra. This results in the spectra which are of reasonable quality as seen in our raw data.
3. The references provided in the manuscript to the earlier work carried out with commercial accessories described differences between the solution spectra and the solid state spectra and list changes in both intensity and λmax shift. In order to validate our method, we wanted to point out that both/similar changes occur with our method as well. However, as Table 1 of our manuscript shows, we have focused on the peak position. In the amended manuscript we will point out that intensities can be affected by many variables and that one should rely more upon the λmax position. These explanations will be added to our amended manuscript.
1. The work is not aimed at suggesting that fluorescence emission spectroscopy should be applied to solid samples of proteins instead of using their solutions. On the other hand, as discussed in the manuscript, there are many investigations (enzyme immobilization on solid surfaces, enzyme powders in low water enzymology) wherein structural characterization would be valuable with the solid state samples. The availability of some commercial accessories fulfills this need. So, we offer a way of measuring fluorescence spectra in a 96-well format without any additional accessories.
2. The 96-well plates used were black from all sides except from the top. So, the emission takes place along the same path direction as the exciting radiation. This minimizes scattered light and distortion of the spectra. This results in the spectra which are of reasonable quality as seen in our raw data.
3. The references provided in the manuscript to the earlier work carried out with commercial accessories described differences between the solution spectra and the solid state spectra and list changes in both intensity and λmax shift. In order to validate our method, we wanted to point out that both/similar changes occur with our method as well. However, as Table 1 of our manuscript shows, we have focused on the peak position. In the amended manuscript we will point out that intensities can be affected by many variables and that one should rely more upon the λmax position. These explanations will be added to our amended manuscript.
Competing Interests:No competing interests were disclosed.Close
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)