Keywords
distant relationships; function annotation; sequence search algorithms; protein superfamilies
distant relationships; function annotation; sequence search algorithms; protein superfamilies
We have now modified the title in response to comment from Dr. Chakrabarti to emphasize that it is a search approach not an algorithm. In such repetitive sequence searches, the best improvement is not only in high coverage but in achieving it with minimal computational time. In this respect, BRS-MP is recommended if the searches are likely to be exhaustive with multiple queries. Likewise, we mention recommended best strategies, such as gathering homologues at 60-90% and to retain motifs less than 15 residues, as suggested by Dr. Madhusudhan. These are now mentioned in the Conclusions. Since the search is against the NR sequence database, there is no firm gold standard to recognize false positives (point 3 made by Dr. Saikat Chakrabarti) and hence it is hard to compare the performance using traditional ROC curves. In response to comment from Dr. Madhusudhan, we have now included one more example on a protein superfamily. MQ or MPMQ approach, whilst being obvious choices, are also computationally demanding and this analysis on 12 superfamilies was a conscious attempt to measure the extent of gain/loss of coverage. We have now introduced a Venn diagram that provides a cumulative measure of coverage of these different approaches (Figure 4-A of the revised manuscript). We do find similar trends when the improved sequence search approach is applied on 1961 superfamilies.
See the authors' detailed response to the review by Saikat Chakrabarti
See the authors' detailed response to the review by Mallur Srivatsan Madhusudhan
Protein sequence databases have grown enormously in recent times. Understanding protein homology within such huge sets of sequences requires tracing the divergence by mutation, substitution, insertion and deletion of residues1,2. Homologous proteins reflect similarity at sequence and structural levels, implying functional similarity3. This level of similarity broadens into the superfamily and the ways to deduce such relationships differ for both protein sequence and structure information4,5. There are different databases that organize sets of homologous proteins or protein superfamilies based on protein sequence and structure. These databases primarily employ protein domain information present in a sequence or structure. SCOP is a database that organizes the protein structural domain data in different hierarchical levels based on structural and functional information6. Many sequence search strategies use SCOP domains as a starting point for homology detection, focusing mainly at the superfamily level7,8. Structure-based classification is helpful to explore sequence space and helps in functional assignments by association of protein sequences9.
Of the several methods developed for protein homology detection, the popular BLAST10 algorithm uses heuristics to search sequences and is able to detect close homologues, but fails in a few instances to establish relationships between distantly related proteins. To detect remote homologues, several methods such as, PSI-BLAST10 based on profiles, Hidden Markov Model (HMM)-based methods like HMMSEARCH and Jackhmmer11,12, pattern-based methods like PHI-BLAST13, intermediate sequence search methods such as Cascade PSI-BLAST14 and phylogenetic tree based searches like Treesearch15 have been developed. Methods such as CHASE incorporate some of the above methods in a combined manner to reinforce the sequence search16. Each of the above methods can be optimized for better performance by customizing their parameters and the way they are implemented for sequence searches. For instance, PSI-BLAST is an iterative PSSM (Position Specific Scoring Matrix)-based remote homology detection method. In Cascade PSI-BLAST the search is iterated for several generations in a cascaded manner to improve remote homology detection14. It is important to select appropriate start points for sequence searches, especially for protein families and superfamilies, as different start points can result in different coverage. Park and co-workers have shown that remote homology detection is enhanced threefold for a set of related sequences in the form of a profile than merely searching with a single sequence as a query17. Anand and co-workers emphasized the use of multiple PSSMs as better detectors of remote homologues compared to a single query18. Thus, a search strategy can be designed to improve remote homology detection by choosing appropriate method(s) and starting point(s) for the search and by further optimizing the parameters.
We have considered multi-member superfamilies from the PASS2 database19. PASS2 is a database of structural alignments of protein domains in a SCOP superfamily which share less than 40% mutual sequence identity. The strategy lies in using all the members of PASS2 superfamily, i.e., multiple members, and searching against the NR-Db (Non Redundant Database) available at the NCBI protein resource. We present our analysis based on a multiple query approach (MQ) for PSI-BLAST and PHI-BLAST. While using PHI-BLAST, for each query we obtain a set of patterns to initiate multiple searches per query, thereby adding an extra dimension for multiple patterns. The search approaches are evaluated at different coverage levels and a comparison of the two approaches using the two methods is presented. The methods are then applied to the entire PASS2 database.
Different structural classes of proteins described in SCOP (version 1.75) were considered for remote homology detection. PASS219 based on the SCOP database and the ASTRAL compendium20, uses protein structural entries from the SCOP superfamily with less than 40% mutual sequence identity. Three superfamilies each from four structural classes from the PASS2 database (2008 version) were selected for this study (Table 1). Each superfamily contains a varied number of members, with some containing a single family and others with more than one family. The classification of families is based on SCOP hierarchies; PASS2 does not consider this classification. Therefore, different members within a PASS2 superfamily may be listed in different families in SCOP. The sequence search strategy devised, described below, was implemented for data mining the sequence homologues for structural superfamilies. Structural members from all the 1961 PASS2 superfamilies were used to search for sequence homologues.
Each member of the PASS2 superfamily was selected as a query for homologue detection and used to search against the NR-Db available at the NCBI ftp site (ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz). Two popular methods, PSI-BLAST and PHI-BLAST, from the BLAST 2.2.23+ package, were used for sequence searches (ftp://ftp.ncbi.nih.gov/blast/executables/blast+/2.2.23/ncbi-blast-2.2.23+-x64-linux.tar.gz) (see Figure 1 for flow-chart).
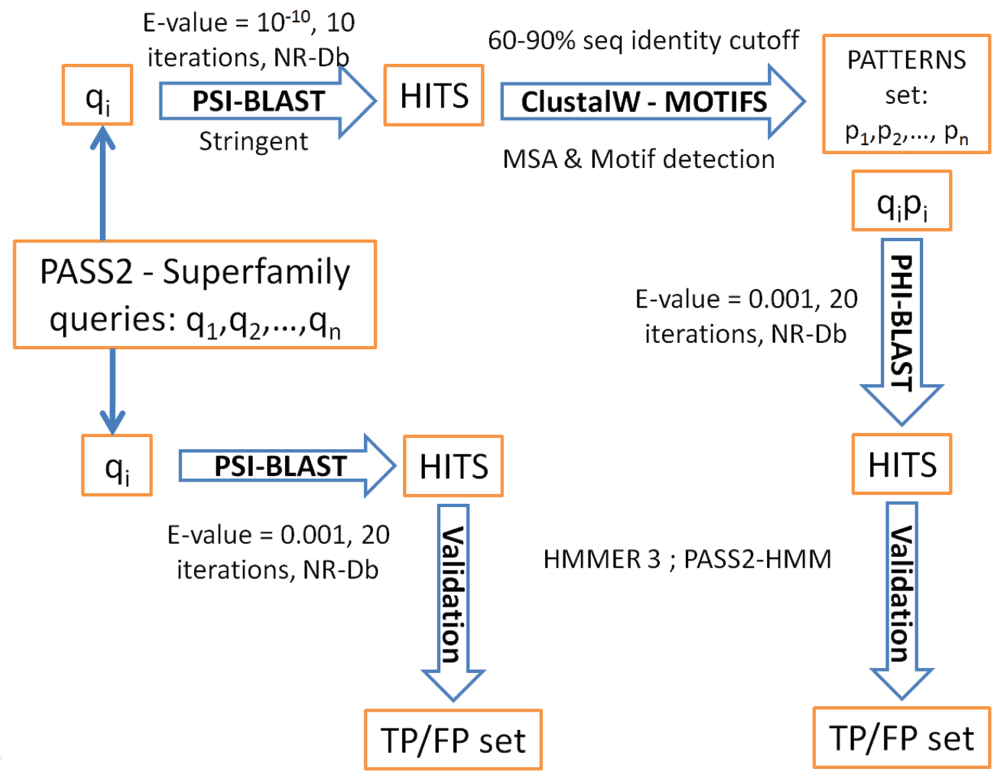
PSI-BLAST was used for each query until convergence or a maximum of 20 iterations. The expectation value threshold (E-value: parameter –e) and inclusion threshold (parameter –h) were both set to 0.001. In all the iterations, all the alignment regions from each of the hit sequences were considered for further validation steps. Following this, a set of all unique sequence homologues (hits with a unique GenInfo Identifier (GI)) was recorded for each member. Sets of hits from all the members in a superfamily were pooled and filtered to generate a cumulative set of unique GIs for each superfamily. Employing all members for homology detection for a superfamily is essentially the multiple query (MQ) approach in the sequence search strategy. For all the PASS2 superfamilies, data mining was carried out using this MQ approach and PSI-BLAST on the latest NR-Db version (June, 2012 release) to populate a sequence homologue database for structural superfamilies7. PHI-BLAST requires a query and a pattern pair as an input. A set of patterns was identified for each query and each query-pattern pair was used for the PHI-BLAST search. The parameters and their values were maintained as in PSI-BLAST searches. The use of multiple patterns for all the multiple queries (MPMQ approach), for superfamily level homology detection was tested on the 12 PASS2 superfamilies.
Homologues were searched for query sequence patterns, using stringent PSI-BLAST criteria (maximum 10 iterations, 10-10 E-value), in NR-Db. A set of hits with 60–90% sequence identity was selected for the pattern generation process. For queries with <10 hits, the sequence identity window was relaxed to 40–95% so that there were enough sequence homologues to deduce a conserved pattern. These hits were aligned using ClustalW 2.021. An in-house program, MOTIFS (the program can be made available upon request), was used to identify motifs from the Multiple Sequence Alignment (MSA). The program accepts a MSA of protein sequences as input and employs a DAYHOFF-type amino acid exchange matrix to score amino acid replacements for every pair of sequences. The average pairwise amino acid exchange scores are recorded at every alignment position. In general, an alignment position with a high score means a highly conserved position. A motif is reported for a stretch of three or more consecutive positions with complete conservation or favorable substitution (based on an identical or similar residue in a given position in the MSA and the average score). For all such identified motifs, the stretch of amino acids from all the sequences is reported with their length and position corresponding to the first sequence in the alignment. The motif generated is screened for the following conditions:
Length of the motif should be ≥ 3
At each position, there should be only ≤ 3 different amino acid preferences (reported by MOTIFS); if not, then that position is denoted as ‘X’, where ‘X’ refers to any amino acid at that position
When there is an occurrence of two consecutive ‘X’s (variable residues), the pattern is divided into two separate patterns
No pattern starts or ends with ‘X’
Any pattern can have a maximum length of 15 amino acid positions, since above this length there is little increment in the number of hits obtained. If length of a pattern is >15 amino acid positions, it is broken into subsets of patterns which meet all the above conditions and has a maximum length of 15.
After the motif passes through all the above filters, it is converted into a PROSITE format pattern, and used for PHI-BLAST along with its query22.
The PASS2 database contains structure-based sequence alignments which are used to build a HMM for each superfamily. Since these HMMs are built using structural alignments, they are highly sensitive in validating the sequence homologue obtained for a superfamily member as a true positive (TP). A library of all PASS2 superfamily HMMs was consulted using HMMSCAN, from the HMMER 3.0 package (ftp://selab.janelia.org/pub/software/hmmer3/3.0/hmmer-3.0-linux-intel-x86_64.tar.gz), to validate all the sequence homologues through either of the sequence approaches (MQ and MPMQ) discussed above11. This approach of validation had earlier been effective in 80% of the examples23.
In the process of data mining for all PASS2 superfamilies using the MQ approach, additional validation was used for the homologues which failed to associate with the correct superfamily HMM. A single query HMM was built for all PASS2 members which were grouped together to generate a library. Thus, an additional validation using single-query HMM was carried for all PASS2 superfamilies. All the HMMSCAN runs were performed with an E-value threshold of 0.01.
Coverage for both of the sequence search approaches was based on different levels. Firstly, a cumulative set of validated hits (Cum-TP) obtained for each superfamily should contain all the PASS2 members comprising that particular superfamily. Secondly, hits corresponding to the sequences of all the SCOP members for a given superfamily should be part of the Cum-TP set. Since, PASS2 contains superfamilies where no two members within a superfamily had >40% sequence identity, the coverage at the SCOP level reflects the sensitivity of the search strategy. Any cross-superfamily hits detected were considered as false positives (FP), even if they belong to the same fold. Thirdly, since sequences of all structural entries present in Protein Data Bank (PDB) obtained as hits are not classified into SCOP, all structural entries were inspected through HMM validation whether they associate to a single PASS2 superfamily HMM with no cross-superfamily associations. Finally, all the protein sequence homologues devoid of the above mentioned levels, with no structural information, were validated by HMMSCAN.
In each superfamily, a set of cumulative true positives (Cum-TP) was formed with hits having unique GI obtained from different members. Therefore, from the set of all hits obtained for a superfamily (All Hits), the number of validated hits, i.e. Cum-TP, was used to calculate positive prediction value (PPV) for all 12 superfamilies in both approaches. The PPV is calculated as follows:
A member with highest number of TPs was identified as a best representing sequence (BRS) and its TP count was also recorded (BRS-TP) for all superfamilies. Corresponding to Cum-TP, a ratio of BRS-TP/All Hits was also calculated. Using this value and Cum-TP, a percentage gain in the coverage (PGC) was recorded:
The overlap between coverage achieved by the four cases – MPMQ approach, MQ approach, BRS from PHI-BLAST and PSI-BLAST was assessed by plotting a Venn diagram24. Following these fourcases, all the validated hits obtained for all the 12 superfamilies were segregated into 4 sets and used for plotting the Venn diagram.
The sequence search strategy devised for remote homology detection was tested and implemented.
Selection of parameters. 12 structural superfamilies were considered for testing the sequence search strategy. These superfamilies span different classes (α, β, α/β, α+β) of proteins, as per the SCOP definitions. In the MQ approach, all the members from each of the 12 selected superfamilies were used as inputs for PSI-BLAST to search against the NR-Db. The performance of PSI-BLAST was optimized for different parameters. The E-value (parameter –e) and inclusion threshold (parameter –h) were optimized to 0.001, after testing it on a range of 1 to 10-10. An optimized E-value ensures better coverage with fewer FP. The number of iterations (parameter –j) was set to 20, after testing for values 5, 10 and 20. Some queries may converge within 5 iterations where the superfamily was less diverse as in the case of 50203 (Bacterial enterotoxin). However, for certain superfamilies like 55239 (RuBisCo-small subunit), none of the members could converge searches within 20 iterations explaining the abundance of sequence homologues for such proteins in the NR-Db. The rest of the parameters were set to default.
Query retention. In the profile-based iterative (PSI-BLAST) run, the sensitivity and specificity depends upon the quality of the PSSM generated per iteration. If the PSSM gets corrupted, then the PSI-BLAST search drifts to FPs by inclusion of non-homologous sequences. This can be traced by retention of the query in the PSSM throughout the iterations of a PSI-BLAST run. A query retained until the end reflects good optimization of search parameters and little corruption of the PSSM. To study the query retention for each member in a superfamily, the presence of query was inspected in all iterations for the PSI-BLAST run. The number of iterations was divided into four bins as 25%, 50%, 75% and 100% of total iterations for which the PSI-BLAST run lasted or converged. If a PSI-BLAST run converged at the 16th iteration, upper limits for these four bins were 4th, 8th, 12th and 16th iterations, respectively. The query was inspected for the occurrence in any of the four bins for all members of a superfamily. The frequency of queries last observed in any of the four bins is plotted in Figure 2-A. It reflects that out of a total of 70 queries from all 12 superfamilies, 45 queries were retained until the last iteration and 23 queries were not retained beyond 50% of iterations. However, it was observed that close homologues of the member query (sequence identity >90%) were retained until the last bin, thereby driving the PSI-BLAST search in the correct direction. Query drift was observed in only two out of the total 70 members from 12 different superfamilies.
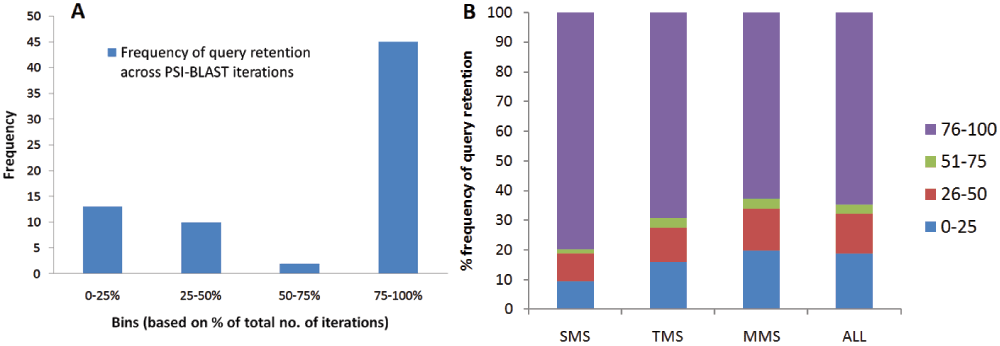
(A) Number of member queries observed per bin for 12 superfamilies and (B) A stacked bar plot for all percentage query retention bins observed for all superfamily members in PASS2 database and also for the different sets of SMS (Single-Member Superfamily), TMS (Two-Member Superfamily) and MMS (Multi-Member Superfamily).
In the data mining of all structural superfamilies within the PASS2 database using the MQ approach, a similar trend was observed for the entire set of 10569 queries arising from 1961 superfamilies. 65% of queries were observed in the fourth bin, thereby ensuring the parameters selected for this scale-up of a MQ approach were acceptable. At times, the loss of query in the PSSM could still be beneficial, if the resultant PSSM is enriched by neighboring families, thus improving the coverage at the superfamily level. The PASS2 database includes the classification of superfamilies as single-member (SMS), two-member (TMS) and multi-member (MMS) based on the number of members in each superfamily. A distribution for the query retention in the four bins for these sections is shown in Figure 2-B. 68% of MMS queries have >50% query retention. However, in the MQ approach, few of the members in MMS are retained until the fourth bin to ensure better sequence search at the superfamily level.
Each member and pattern pair was used for sequence searches using PHI-BLAST. Similar to PSI-BLAST, the E-value (parameter -e) and inclusion threshold (parameter -h) were optimized to 0.001. The maximum number of iterations allowed was set to 20. While generating the patterns, it was ensured that patterns are specific and stringent to query. The length of the patterns generated varies from 3 to 40 amino acid positions; however during sequence search using PHI-BLAST, the pattern length of maximum 15 amino acid positions was used. Longer lengths reflect the long conserved stretches in the homologues selected for pattern generation. Multiple patterns for each query and multiple queries per superfamily (MPMQ approach) were used in the sequence searches. This gives rise to multiple start points to search in sequence space for each superfamily. Although PHI-BLAST essentially follows an iterative protocol like PSI-BLAST, the hits identified in the first iteration differ as they are constrained to have the pattern specified along with the query. This ensures that the hits will be used to create the first PSSM for the rest of the iterative search process.
The sequence hits obtained through MQ and MPMQ approaches were validated for TP and then inspected for the coverage at different levels for all 12 superfamilies.
Coverage for PASS2 members. Every member was examined for its coverage of all the PASS2 members in the sequence search. All the members achieved full coverage when searched in the cumulative set of hits derived from sequence searches of all the PASS2 members. Every member of the two superfamilies, RuBisCo small subunit (SCOP code: 55239) and mannose-binding lectins (SCOP code: 51101), could identify all other members. But for the rest of the superfamilies, every member was unable to identify all the PASS2 members. Although, single members in such instances were unable to cover all the PASS2 members cumulatively, presence of all members gave rise to 100% coverage, thereby stating the importance of the use of multiple queries (MQ).
Coverage for SCOP superfamily members. The coverage for all the SCOP superfamily members was inspected. The ratio of the number of sequence homologues of SCOP superfamily entries identified as TP to the total number of SCOP superfamily members is plotted as shown in Figure 3 (blue bars). Almost all of the superfamilies obtained >90% coverage for all SCOP superfamily members. The only exception was the Bacterial enterotoxins superfamily with two families (SCOP code: 50203), where coverage was 25%. In the PASS2 database, this superfamily contained nine members from one SCOP family, and could identify most of its SCOP family members. But none of the members could identify cross-family members. However, there was only one member from the other family which was not sufficient to identify all the family members. This lack of coverage can be overcome if more members are included from the second family. For such a diverse superfamily, it is apparent that unless we use an MQ approach, it is difficult to achieve a reasonable coverage. In the rest of the superfamilies, some members were able to identify cross-family members while some could not identify any cross-family member. In either case, all of the SCOP family members were covered. Cumulatively, all PASS2 members are strong enough to cover most of the SCOP superfamily members underlining the high sensitivity of MQ approach.
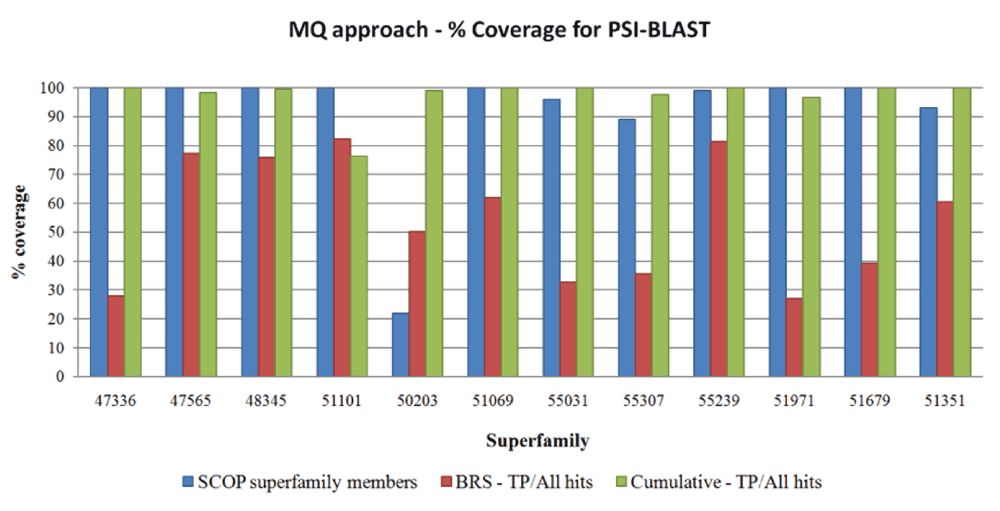
The blue bars correspond to the percentage of SCOP members identified as hits. The red bars correspond to percentage ratio of TP (True Positives) identified by BRS (Best Representative Sequence) and All Hits (BRS-TP/All Hits). The green bars correspond to percentage ratio of Cumulative TP identified for a superfamily and All Hits (Cum-TP/All Hits).
Coverage for the structural (PDB) entries. The sequences of structural entries present in PDB were part of the All Hits set. These are newer entries which are yet to be accounted in the SCOP database. After PASS2-HMM validation, they were inspected for which superfamily they belong to and if they could associate to the query member’s superfamily. There were no cross-superfamily connections, highlighting the specificity of the sequence search through the MQ approach.
Coverage with respect to the TP sequence homologues. While following the MQ approach over a huge database like NR-Db, it is necessary to achieve a maximum number of homologues (TP) and minimum unrelated sequences (FP). The use of multiple queries ensures a high TP count, while appropriate stringency of the E-value restricts occurrence of FPs. Following validation based on PASS2 HMMs, a set of Cum-TP was obtained and a ratio of Cum-TP/All Hits was calculated. Similarly, a BRS-TP/All Hits ratio was also calculated. Both these values are expressed as percentages as shown in Figure 3 (green and red bars respectively). The percentage of SCOP entries identified is also represented in Figure 3 for each superfamily. Considering the Cum-TP/All Hits ratio, Figure 3 shows the extent of coverage achieved by the BRS. It is clear that none of the BRS could achieve more than an 80% TP share of all the TPs identified at superfamily level. The average coverage for BRS across all superfamilies was ~50%. However, the Cum-TP/All Hits ratio was >90% for most of the superfamilies. In most of the cases, the false positives were hits which could not get associated with any PASS2-HMM, but did not associate with any other superfamily apart from query superfamily. This may partly be due to the fact that the superfamily was so diverse that the PASS2-HMM itself was getting noisy due to the diversity or else the hit obtained was too distant to get validated using the HMM validation cut-offs. On average, false positives were <3%. The results strongly suggest that the MQ approach to sequence data mining helps to increase coverage at the complete superfamily level. For superfamilies which have multiple families (47336, 55031), none of the BRS could obtain a BRS-TP/All Hits ratio >35%, but cumulative coverage by all queries for a complete superfamily is excellent (100% Cum-TP/All Hits ratio). Even in the case of a single-family superfamily (55031 - Bacterial exopeptidase dimerisation domain superfamily), all the members have attained a 100% Cum-TP/All Hits ratio.
The coverage of the MPMQ approach for all 12 superfamilies was equal to or better than the MQ approach at each level discussed above. All the PASS2 members were identified cumulatively by all the members. The coverage increased for all the superfamilies with respect to the SCOP superfamily (except for the 50203 superfamily which performed poorly even in the MQ approach). The sequences of structural (PDB) entries identified were associated with the query superfamily PASS2-HMM. Similar to PSI-BLAST, PHI-BLAST results also revealed that the use of multiple queries are beneficial for achieving better coverage at PASS2, SCOP and PDB level.
The MPMQ approach was contrasted with the MQ approach (Table 2). The high PPV value suggests that both approaches were able to discern between the TP and FP. The MPMQ approach has relatively more start points than the MQ approach, employing pattern-constrained PHI-BLAST. The BRS identified in PSI-BLAST was also the same in the PHI-BLAST results for most of the superfamilies. However, the performance of BRS from PHI-BLAST using multiple patterns (MP) was even better for some of the superfamilies than MQ approach wherein all the queries were used through PSI-BLAST for sequence search. There was an increase in the number of TP identified for BRS PHI-BLAST as against MQ in 5 out of 12 superfamilies. The TP count for all 12 superfamilies was highest for MPMQ with better PPV for 10 out of 12 superfamilies than MQ approach.
Corresponding to the four approaches – MPMQ, MQ, BRS PHI-BLAST and BRS PSI-BLAST, the total hit count with MPMQ was highest for each superfamily. Hence, all the hits obtained were combined for all 12 superfamilies together. Following the above mentioned four approaches, the total hits-set (36111 hits), was segregated into four sets (MPMQ = 35680, MQ = 22744, BRS PHI-BLAST = 16909, BRS PSI-BLAST = 9079) and plotted as a Venn diagram (Figure 4-A). The total hit sets obtained by BRS of both PSI-BLAST and PHI-BLAST were subsets of the MQ and MPMQ approaches, respectively. However, it was interesting to find that 98% of the hits identified through MQ approach were covered by MPMQ, emphasizing the performance of MPMQ over MQ approach. Moreover, MPMQ approach covered 37% more hits which were not identified by MQ approach. The BRS PHI-BLAST approach alone was able to identify 15% more hits than MQ approach, suggesting that employment of BRS PHI-BLAST approach alone can augment the MQ approach for a gain in coverage.
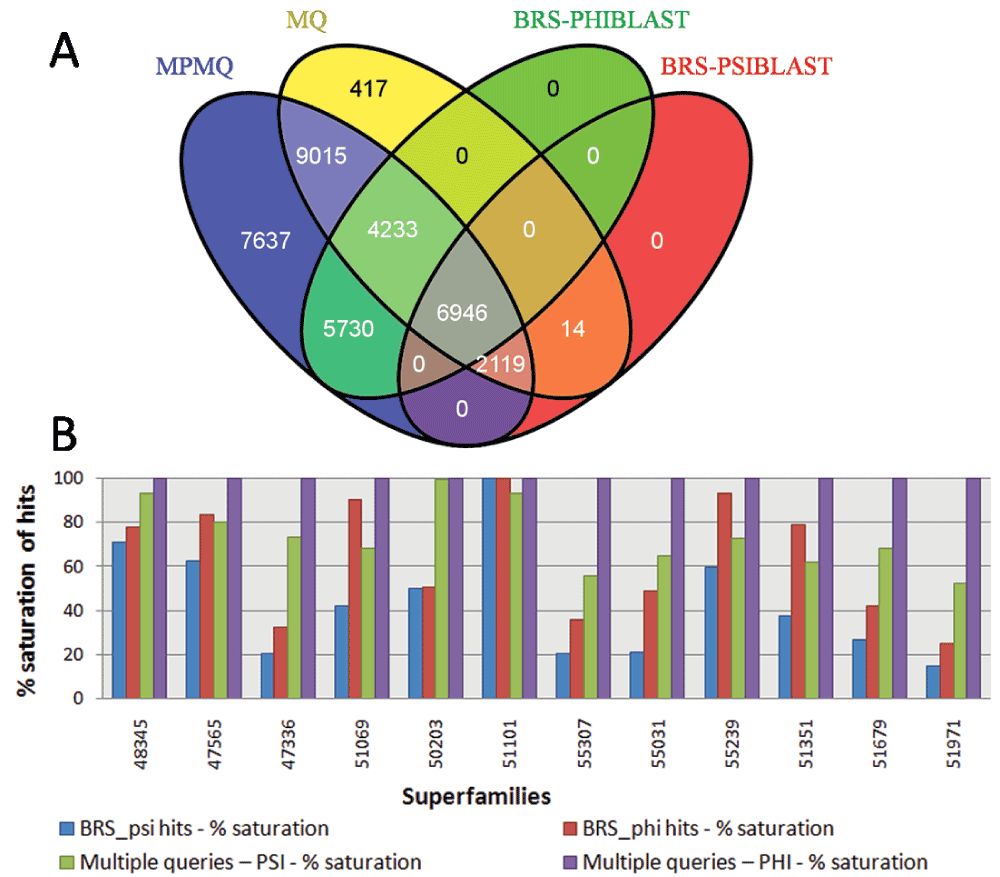
(A) Venn diagram representing four sets MPMQ (Multiple Pattern-Multiple Query), MQ (Multiple Query), BRS (Best Representative Sequence) from PHI-BLAST and BRS from PSI-BLAST comprising of all hits combined for 12 superfamilies. (B) Comparison of MPMQ and MQ with respective BRS-TP (Best Representative-True Positives). The blue and green bars correspond to TP obtained for BRS (BRS-TP) and cumulatively (Cum-TP) using all queries in the superfamily respectively using PSI-BLAST. The red and purple bars correspond similarly to TP obtained for BRS (BRS-TP) and cumulatively (Cum-TP) for all queries in the superfamily respectively using PHI-BLAST. All values are % saturation considering Cum-TP for PHI-BLAST (highest number of TP) as the saturation value for each superfamily.
The MPMQ approach covers 99% of the total hit set. This was chosen as 100% coverage (% saturation value) to compare performance of rest of the 3 cases (viz. MQ approach, BRS PHI-BLAST and BRS PSI-BLAST) for each superfamily, separately. The performance was compared by plotting percentage coverage achieved using the BRS-TP/All Hits and Cum-TP/All Hits (Figure 4-B). The Cum-TP/All Hits was highest in all superfamilies for the PHI-BLAST runs. This was chosen as 100% coverage (% saturation), to compare performance of BRS in MQ and MPMQ and Cum-TP/All Hits for the MQ approach. The Cum-TP/All Hits (MPMQ) was almost double for 55307 and 51971. This difference between MPMQ and MQ approaches was >20% for 9 out of 12 superfamilies. Although the MPMQ approach has promising results in comparison to the MQ approach, it is computationally very expensive. Each member in a superfamily generates multiple patterns, hence the higher the number of query-pattern pairs means the number of PHI-BLAST runs to be performed for the given superfamily increases proportionally.
The performance of BRS in both approaches was also inspected. BRS for MPMQ achieved close to 80% coverage in 9 out of 12 superfamilies, with an exceptionally low value of 25% for the 51971 superfamily (a multi-family superfamily). Considering multi-family superfamilies (47336, 50203, 51679, 51971), the BRS of MPMQ could not cross 50% coverage compared with the Cum-TP/All Hits of MPMQ. This reinforces and lends support to the strategy of using multiple queries for sequence searches at the superfamily level. The MPMQ BRS was able to identify more TPs than Cum-TP obtained through MQ for the 51069, 55239, 51351 superfamilies, which are single-family superfamilies. This implies that BRS in isolation, through PHI-BLAST, can drive sequence searches attaining better coverage in such superfamilies, achieving a trade-off between coverage and computational time. However, performance of BRS is highly superfamily-specific.
GenDiS (Genomic Distribution of protein structural domain Superfamilies) is our previous database of sequence homologues based on PASS2, where sequence homologues for structural members are organized with respect to their genomic distribution at the superfamily level7. In an attempt to populate GenDiS, the MQ approach was implemented to search in sequence databases starting with all the 1961 PASS2 superfamilies. PASS2 classification of SMS, TMS and MMS relies on the number of constituent members in a superfamily. For each superfamily a percentage gain in coverage (PGC) value due to improvised strategies, in comparison with simple PSI-BLAST runs, was calculated based on the preliminary results on 731 MMS (see Methods). The PGC was divided into four quarters and the frequency of superfamilies belonging to each quarter is shown in Figure 5-A. It was found that 532 superfamilies obtain >50% PGC. This observation strongly suggests the necessity of the MQ approach for remote homology detection, where the BRS alone collects fewer homologues. The MQ approach has to be implemented, especially for huge databases like NR-Db, to obtain improved coverage at the superfamily level. An inspection of the relation between number of members and PGC for MMS is shown in Figure 5-B, revealing an increase in PGC value with a rise in the number of members. The PGC values are extremely high for diverse and large superfamilies with more than 30 members.
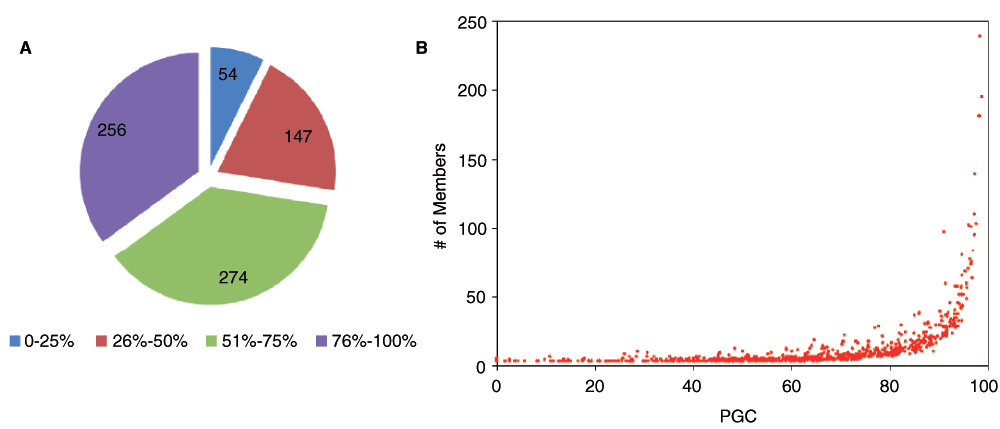
(A) Frequency of superfamilies based on PGC (Percentage Gain in Coverage) value divided into 4 bins (0–25%, 26–50%, 51–75%, 76–100%). (B) The PGC value plotted against number of members for all superfamilies.
We have employed structural entries from the PASS2 database to understand and improve sequence data mining techniques, where the searches are specifically meant for detection of distant homologues19. SCOP is a good benchmark dataset to evaluate many homology detection methods6, but removing redundancy with respect to similar entries reduces the search time. However, with the use of the MQ approach for searching sequence space there is substantial improvement in coverage, albeit with an increase in search time. Use of PASS2 superfamily members as query is a trade-off, between use of all the SCOP members as query (ideal for best coverage) and a single query for the entire structural superfamily (ideal for saving computational time).
MQ grants an additional advantage of coverage, which is revealed by considering coverage at different levels. A multi-member SCOP superfamily like 48345 (a virus capsid protein alpha helical domain), with multiple families (Orbivirus capsid [SCOP code: 48346], Phytoreovirus capsid [101395] and vp6, the major capsid protein of group A rotavirus [63596]), where each query gives an exclusive set of hits for each query (representing its SCOP family) through both the methods, use of multiple queries is inevitable to achieve a better or even complete coverage. Similarly, another multi-member, multi-family superfamily 51971 (Nucleotide-binding domain) contains nine PASS2 members divided among three SCOP families (N-terminal domain of adrenodoxin reductase-like [SCOP code: 51972] - five members, D-aminoacid oxidase, N-terminal domain [51979] - two members and UDP-galactopyranose mutase, N-terminal domain [69427] - two members). In this superfamily, PASS2 members of one family could not identify PASS2 members from other family. Owing to the diverse nature of this superfamily, it is necessary to adopt the MQ approach for achieving better coverage. Further, for this superfamily, MPMQ approach provided multiple start points and a two-fold increase in coverage as compared to MQ approach. However, it was not possible for PASS2 members to establish a cross-family connection. Therefore, choice of multiple queries with inclusion of their respective multiple patterns increases the overall coverage for diverse superfamilies. These findings comply with previous findings based on different search methods17,25.
To date, many methods and approaches have been standardized for sequence searches at different levels of database complexity8,16–18,25. PSI-BLAST is in many instances a primary choice for homology detection. Efforts by different groups have enhanced the usage of PSI-BLAST, improving its sensitivity in remote homology detection14,26. PHI-BLAST, employing an initial pattern–hit initiation, enhances the search method before continuing over profile-based searches in subsequent iterations25. In the present study this has been shown to increase the coverage by >20% for many of the selected superfamilies. The simultaneous use of different methods greatly improves coverage, and the combinations of other methods needs to be investigated.
The choice of BRS identified from MQ may provide a better option to run PHI-BLAST for a single query. This is beneficial considering an increase in coverage, but it is necessary to acknowledge that, multiple queries and their multiple patterns increase computational time tremendously for a given superfamily. Although for a multi-member, multi-family superfamily, this may not effectively work, this approach can serve as a good trade-off between coverage and computational time for a superfamily with a single family. Also, for a very large superfamily (>20 members), the BRS-PHI-BLAST will be a good option to choose, however the choice of any of the discussed approaches for sequence search becomes superfamily specific.
In the post-genomic era, there is a strong need to devise sequence search strategies for effective functional annotation. Functional annotation transfer and convincing establishment of sequence relationships are bottlenecks at low sequence identities. Hence, there is a constant quest for a robust computational program which is greedy on coverage and stringent in eliminating false positives. Remote homology detection through sequence searches has been addressed by various methods and approaches. Considering a protein structural superfamily, when an attempt to view its spread in the sequence space is made, one can resort to various methods and approaches. This study shows that a MQ approach proves beneficial for increasing the coverage. The PASS2 database, which accounts for SCOP superfamily members that are structurally aligned, becomes a good start point to scan sequence space for good coverage at the complete SCOP superfamily level. Instead of using all SCOP members, using only PASS2 members with a MQ gives rise to good coverage in remote homology detection. In this paper, we have further compared the MQ approach using PSI-BLAST to a MPMQ using PHI-BLAST. The MPMQ approach totally outperforms the MQ approach. Use of patterns makes the initial pattern-hit initiation stringent, thus elevating the specificity of the search25. The study also indicates that a best representative sequence performs better with the PHI-BLAST than the BRS PSI-BLAST while better or equivalent to the MQ PSI-BLAST approach for certain superfamilies. Therefore the use of BRS PHI-BLAST can help save computational time and achieve better coverage. But this is superfamily-specific and for multi-family superfamilies, a query from one family may not be sufficient to associate with hits obtained from a query of a different family within a given superfamily. It is, therefore, necessary to resort to a MQ or MPMQ approach for getting reasonable coverage. The PGC obtained from data mining for all multi-member superfamilies from PASS2 points to the necessity of using multiple queries. The MQ approach is the easiest to follow for an end-user to achieve good coverage at superfamily level, while using MPMQ approach will improve the extent of sequence coverage. However, if there are many members (>10) in a superfamily, MQ approach alone can be adopted. Alternatively, for representative sequences, multiple pattern based PHI-BLAST can be used to achieve a trade-off between computational time and coverage. Thus, different methods and different approaches can be used to improve the sequence searches during remote homology detection.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)