Keywords
17β-estradiol, breast cancer, apoptosis, IKKα, BAY11-7082
17β-estradiol, breast cancer, apoptosis, IKKα, BAY11-7082
Our research projects currently focus on understanding of the interplay between the different signaling cascades and estrogen receptor (ER) dependent transcription activation in breast cancer. For many years, those of us working in the field were used to looking at estrogen as a mitogen through its genomic function mediated by the ER-dependent transcription program. Now we realize that estrogen through ER, in addition to regulate gene expression, crosstalks with many non-genomic signaling pathways involved in cell growth, differentiation and apoptosis. These newly identified interplays may give a different flavor to long conceived mitogenic role of estrogen. In this opinion article, we generally reviewed the changing attitude toward estrogen in clinical use, with a focus on a new discovery that estrogen, in combination with IKKα, can induce breast cancer cell apoptosis effectively. We also discuss the possibility of estrogen and IKKα inhibitor dual-therapy strategy in cancer treatment.
Sir George Thomas Beatson (1896) used oophorectomy to reduce the estrogen level in premenopausal women in order to prevent breast cancer occurrence, and he was the first to reveal the relationship between estrogen levels and breast cancer1. Half a century later, Haddow et al. (1944) first used a high-dose synthetic estrogen (stillbestrol) to induce tumor regression in hormone-dependent breast cancer in postmenopausal women2. Huggins et al. (1952) pioneered adrenalectomy to reduce estrogen level for treating mammary cancer3. Huggins’ work is internationally recognized by the prestigious Nobel Prize (1966) for the contribution to the development of endocrine therapy in hormone-regulated cancer. Jensen E (1958) characterized the first receptor for estrogen - estrogen receptor alpha (ERα). Soon after these discoveries, extensive mechanistic studies have gained large information about estrogen’s physiological functions and carcinogenic roles. The Women's Health Initiative (WHI) research program (1991) initiated a 15-year study enrolled 161,808 generally healthy women aged 50–79 to evaluate the beneficial effects of postmenopausal hormone replacement therapy (HRT) on heart diseases, bone fractures, and cancers4. Due to the increased incidence of breast cancer, stroke, and cardiovascular complications in women treated with estrogen alone or with a combination of estrogen and progesterone, the study was terminated prematurely in 2002. Though extensively studied, the definite understanding of the mechanism of estrogen action always challenges our mind.
Estrogen regulates the proliferation and development of tissues expressing estrogen receptors and ERα is mainly expressed in breast epithelium, ovary and endometrium. Thus, estrogen is mitogenic for cultured ER positive breast cancer lines. The mitogenic effects of estrogen at the G1-to-S transition are mediated by the key effectors of estrogen action, c-Myc, cyclin D1 and E2F-15–7. c-Myc expression occurs within 15 min of estrogen stimulation, among the earliest responses to estrogen. Estrogen also rapidly induces cyclin D1 expression. In the G1 phase, estrogen drives E2F-1 expression. Estrogen-triggered all these coherent genetic changes to guarantee the cell cycle progression. Nongenomically, Estrogen binding to the ER stimulates rapid activation of Src and signaling pathways MAPK and PI3K/Akt pathways that affect cell survival8,9. Based on these understandings of estrogen action, ER protein is assayed in newly diagnosed breast cancers because it is a clinically useful prognostic factor and predicts responsiveness to ER blocking drugs such as tamoxifen.
Paradoxically, estrogen induces apoptosis under certain circumstances. As mentioned above, high-dose estrogen was used to induce tumor regression of hormone-dependent breast cancer in postmenopausal women before the introduction of tamoxifen2. This regimen is of clinical interest, given that long-term treatment of breast cancer with anti-estrogen drug tamoxifen often leads to drug resistance and that sustained tamoxifen exposure may sensitize breast cancer cells to high-dose or even low-dose estrogen therapy10. The field of the mysterious dual effects of estrogen on apoptosis have not much progressed until recently.
Recently, Perillo’s Group from Second University of Naples identified a key player, IKKα in the switch of estrogen action in apoptosis11. They found that ER agonist 17β-estradiol (E2) and IKKα kinase specific inhibitor BAY11-7082 (BAY) in combination can induce apoptosis in an ERα-positive breast cancer cell line. Dual-therapy now receives more and more attention.
In the journal Cell Death & Differentiation, Perillo et al. recently reported that the inhibition of IKKα by BAY switched the effect of estrogens on breast cancer cells from anti- to pro-apoptotic, which leads the exploration of therapeutic benefits of estrogen into a new era11. IKKα is the kinase responsible for histone H3 Ser 10 phosphorylation (H3pS10)12. H3pS10 is known to inhibit H3 Lys 9 dimethylation (H3K9me2) in a space repulsion model13. Thus, inhibiting H3pS10 by targeting IKKα facilitates estrogen-triggered ER-dependent recruitment of histone methyltransferase Suv39H1. Histone demethylase LSD1 demethylating the Suv39H1 target sites H3K9me2 was increased concomitantly. LSD1-mediated demethylation process is known to produce reactive oxygen species (ROS) and cause ROS-mediated DNA damaging effects14. The net results after IKKα knowndown is causing more DNA damages to cancer cells through estrogen triggered ER-dependent Suv39H1 and LSD1 binding to ER target gene promoter (Figure 1).
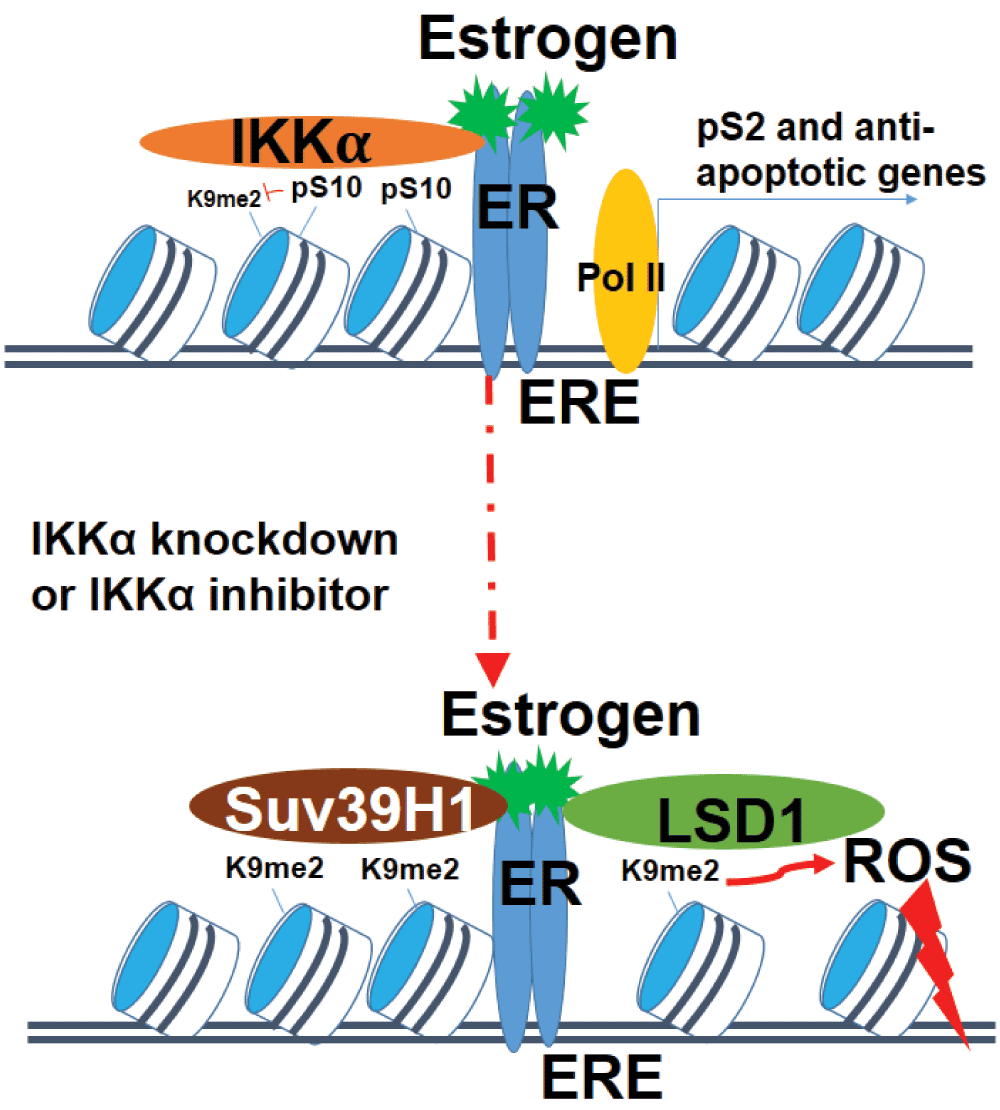
Upper panel: chromatin landscape and factors present at the pS2 (as known as TFF1) promoter in the presence of IKKα. The pS2 promoter is enriched with nucleosomes (blue and white cylinders) that dwell in positions proximal to the transcription start site (+1 position) and at ER binding sites. Only low levels of histone H3 lysine 9 dimethylation (H3K9me2) exist due to the space repulsion of histone methyltransferases binding to H3K9 from IKKα residing at the neighboring H3S10 site. RNA polymerase holoenzyme (Pol II) (yellow oval) is present at the proximal promoter region near the transcription start site (TSS, shown by black vertical line) of the pS2 gene. Lower panel: chromatin landscape and factors present at pS2 promoter following IKKα knockdown or IKKα inhibitor BAY11-7082 treatment. Once levels of IKKα have decreased, ER recruits the histone methyltransferase Suv39H1 or demethylase LSD1 proteins to bind within the pS2 promoter. Once the LSD1 is activated and demethylates its target H3K9me2, it generates reactive oxygen species (ROS) to cause DNA damage effects including base oxidation and nicks results from DNA damage itself and related DNA repair. In sum, the inhibition of IKKα results in the reversion of estrogen triggered anti-apoptotic effects to pro-apoptotic effects.
In short, Perillo's group identifies a novel crosstalk between IKKα and estrogen signaling and shows that inhibition of IKKα-mediated histone phosphorylation switch ER-mediated anti-apoptotic effects to ER-dependent ROS-mediated breast cell death, which implicates potential dual-therapy of ER agonist (E2) together with IKKα inhibitor (BAY) in a variety of hormone-regulated cancers.
In the last few years, there have been significant shifts in the attitudes towards the use of estrogen in clinic. Estrogen exhibits a broad range of functions that regulates cell proliferation and homeostasis in many tissues. Despite beneficial estrogen functions, sustained estrogenic exposure increases the risk and/or the progression of various cancers, including those of the breast, endometrium and ovary15. The International Agency for Research on Cancer (IARC) has listed estrogen as known human carcinogen16. Now, the success of the combination of E2 and BAY will certainly become an “accelerator” to the alternative use of estrogen in treating cancers and we expect to see more positive pre-clinical and/or clinical results in the near future.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)