Perdigones N, Morales M, Mason P and Bessler M. Case Report: Paroxysmal nocturnal hemoglobinuria in a woman heterozygous for G6PD A- [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2014, 3:194 (https://doi.org/10.12688/f1000research.4980.2)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
1Division of Hematology, Department of Pediatrics, Abramson Research Center, The Children’s Hospital of Philadelphia, Philadelphia, 19104, USA 2Division of Hematology, University of Pennsylvania School of Medicine, Philadelphia, 19104-4318, USA
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
We describe a case of paroxysmal nocturnal hemoglobinuria (PNH) in a woman who is heterozygous for the glucose-6-phosphate dehydrogenase A- (G6PDA-) allele. PNH is associated with one or more clones of cells that lack complement inhibition due to loss of function somatic mutations in the PIGA gene. PIGA encodes the enzyme phosphatidylinositol glycan anchor biosynthesis, class A, which catalyses the first step of glycosylphosphatidylinisotol (GPI) anchor synthesis. Two GPI anchored red cell surface antigens regulate complement lysis. G6PD catalyses the first step of the pentose phosphate pathway and enzyme variants, frequent in some populations have been selected because they confer resistance to malaria, are associated with hemolysis in the presence of oxidizing agents including several drugs. The patient had suffered a hemolytic attack after taking co-trimoxazole, a drug that precipitates hemolysis in G6PD deficient individuals. Since both G6PD and PIGA are X-linked we hypothesized that the PIGA mutation was on the X-chromosome carrying the G6PDA- allele. Investigations showed that in fact the PIGA mutation was on the X-chromosome carrying the normal G6PD B allele. We speculate that complement activation on G6PD A- red cells exposed to Bactrim might have triggered complement activation inducing the lysis of G6PD B PNH Type II red blood cells or that the patient may have had a PNH clone expressing G6PDA- at the time of the hemolytic episode.
Corresponding author:
Philip Mason
Competing interests:
No competing interests were disclosed
Grant information:
The work has been supported by the Buck Family Endowed Chair in Hematology, and by NCI NIH grants 2R01CA106995 to PJ Mason, and 2R01 CA105312 to M Bessler.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The major change made in this revised version is that we include a much more detailed case history. When the patient presented to our speciality clinic she had an approximately 6 year history of PNH and for most of the case history we present we rely on the available records. In addition, we changed the following:
We include normal ranges for lab tests.
We have corrected our use of Greek characters in units.
We have included a possible explanation for the hemolysis suggested by one of the reviewers.
We have corrected errors concerning the terms PNH Types I, II and III.
We have corrected the use of the names of co-trimoxazole and eculizimab.
We have added a sentence to Figure 3 legend to indicate the figure is a representative example of our data.
We have included the concentration of DCF used for ROS detection.
The major change made in this revised version is that we include a much more detailed case history. When the patient presented to our speciality clinic she had an approximately 6 year history of PNH and for most of the case history we present we rely on the available records. In addition, we changed the following:
We include normal ranges for lab tests.
We have corrected our use of Greek characters in units.
We have included a possible explanation for the hemolysis suggested by one of the reviewers.
We have corrected errors concerning the terms PNH Types I, II and III.
We have corrected the use of the names of co-trimoxazole and eculizimab.
We have added a sentence to Figure 3 legend to indicate the figure is a representative example of our data.
We have included the concentration of DCF used for ROS detection.
In paroxysmal nocturnal hemoglobinuria (PNH) one or more clones of blood cells develops from stem cells that have an acquired mutation in the X-linked PIGA gene1. The PIGA gene encodes phosphatidylinositol glycan complementation class A, an enzyme that catalyses an early and essential step in glycosylphosphatidylinositol (GPI) anchor synthesis. Thus cells are deficient in all GPI anchored proteins, including CD55 and CD59 which regulate complement activation. PNH usually develops in patients with aplastic anemia (AA) and it is thought that PNH cells have a growth or survival advantage over the AA cells although the mechanism is not known2. PNH cells can be completely deficient in GPI anchored proteins (Type III) or partially deficient due to residual activity of the PIGA protein (Type II), while PNH Type I cells express GPI-linked proteins normally.
Clinically, PNH is characterized by bone marrow failure, thrombosis and intravascular hemolysis. Recently the use of a complement inhibitor, eculizumab has greatly improved the quality of life of PNH patients as it causes a dramatic reduction in the hemolysis and thrombotic episodes, improvement in anemia, with a stabilization of the hemoglobin levels and reduced transfusion requirements3. eculizumab leads to an increase in the number of circulating red blood cells that otherwise are subject to complement-mediated hemolysis4.
Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency is the most common red blood cell enzymopathy and is estimated to affect around 400 million people worldwide5. It is caused by mutations in the X-linked G6PD gene which usually lead to an unstable enzyme. G6PD is needed to maintain NADPH and consequently reduced glutathione levels in red blood cells. G6PD-deficient people, mainly males, can be asymptomatic but are subject to episodes of hemolysis when the red blood cells are subjected to oxidative stress caused by infections, certain drugs or in the case of favism, after eating fava beans6. Several polymorphic variants have been described with specific geographical distributions7. In the African population the most common deficient variant is the G6PD A- variant. Compared with normal G6PD, which is called G6PD B, G6PD A- has two amino acid substitutions Val68Met and Asn126Asp8. These are caused by mutations c.202 G->A and c.376A->G respectively. G6PD A- has a frequency of about 10% in Africans and African Americans. G6PD A differs from G6PD B only by the Asn126Asp change and is electrophoretically distinct but with no significant difference in activity. Though milder than other variants such as G6PD Mediterranean found in Italy, Greece and India, G6PDA- is associated with drug induced hemolysis and patients are advised against taking any substances from a list of those known to cause hemolysis. G6PD deficiency usually only affects hemizygous males and homozygous females but heterozygous females can be affected when, for example, biased X-inactivation has led to a predominance of red blood cells expressing the mutant protein9. Here we present a case of an African American woman who was heterozygous for G6PD deficiency and developed PNH, presenting an opportunity to observe the interaction of these two conditions.
Materials and methods
Peripheral blood from patient CHOP277.01 was obtained after obtaining written informed consent according to the declaration of Helsinki. The Internal Review Board of the Hospital of the University of Pennsylvania approved this study. DNA and RNA were extracted by using QIAamp DNA and RNA Blood mini Kits, respectively, according to manufacturers’ instructions. Blood samples for fluorescent cytometry and electrophoretic analyses were obtained from EDTA tubes and experiments were performed within 2 hours of blood withdrawal.
PCR primers to detect mutations confirming the G6PD A- genotype were designed with Primer3 v4.0. (primers for c.202 G->A mutation: forward 5’- agaagaagatctaccccaccatct-3’ and reverse 5’- ctggtacagagggcagaaccag-3’; primers for c.376A->G: forward 5’-catctgtctgtgtgtctgtctgtc-3’ and reverse 5’- ctcatagagtggtgggaggac-3’). Sanger sequencing was done by the Nucleic Acids core facility at CHOP.
The HUMARA assay was performed as previously described10. Briefly, HhaI digested and non digested DNA was subjected to PCR amplification of the first exon of the HUMARA locus (containing a CAG repeat) using fluorochrome-coupled primers. Amplification products were then migrated on an ABI PRISM 3100 Automatic Genetic Analyzer (Applied Biosystems). Allele calling and the area under the curve (AUC) were determined using GeneMapper v.4.0 software (Applied Biosystems). The AUC was used to calculate the skewing from X chromosome inactivation (XCI). The XCI ratio of the digested fraction was corrected with that of the undigested fraction to allow for preferential amplification of the smallest allele (i.e., the allele containing less CAG repeats). Skewing is present when the percentage of the predominant allele exceeds 74%. A percentage of predominant allele between 90% and 100% is considered extreme skewing.
Measurements of oxidative stress ROS assay was performed as previously described11. Briefly, red blood cells were incubated with 0.4mM 20-70-dichlorofluorescein diacetate (DCF; Sigma) dissolved in methanol. After incubation at 37ºC for 15 minutes in a humidified atmosphere of 5% CO2 in air, the cells were washed, resuspended in PBS and analyzed by flow cytometry (FACSCalibur; Becton-Dickinson, Immunofluorometry Systems, Mountain View, CA, USA). The mean fluorescence channel (MFC) was calculated by FACSDiva software. The identity of the red cell population was verified by staining with an antibody to glycophorin-A. To determine the presence of GPI proteins, cells were labeled with a phycoerythrin-conjugated anti-CD55 antibody. For our experiment, cells from a non PNH- non G6PD individual served as control. The MFC of cells stained with 0.4mM DCF, is proportional to generation of ROS.
The electrophoretic mobility of the protein was performed in cellogel strips as previously described12. Hemolysates treated with and without acidified serum were run in order to assess differences in mobility of the G6PD enzyme.
Case report
A 25-year-old African American woman was referred to the Bone Marrow Failure Outpatient Clinic at the Hospital of the University of Pennsylvania for the evaluation and treatment of her PNH.
The past medical history was significant in that at the age of 19 years she presented to the emergency department with cough and dark urine. She was otherwise previously healthy. Family history was only significant for a sister with sickle cell trait. She was diagnosed with Mycoplasma pneumonia and anemia (Hemoglobin 6.9 g/dL (12–16 g/dL), Hematocrit 19.9% (37–47%)). The anemia was determined to be an autoimmune hemolytic anemia (AIHA; LDH 3170 U/L (87–225 U/L), total bilirubin 1.9 mg/dL (0–1.2 g/dL), indirect bilirubin 1.6 mg/dL (0.2–0.7 mg/dL), reticulocyte count 5.2% (0.5–2.1%)) in the setting of positive IgM cold agglutinin antibodies and positive direct Coombs test. The patient was treated with packed red blood cell transfusions and antibiotics and was discharged. As an outpatient, she was started on steroids, and her hemoglobin stabilized between 9 and 10 g/dL; the cold agglutinin and direct Coombs test became negative. The following year, the patient presented on two separate occasions to the emergency department complaining of abdominal pain and dark urine (urine analysis: RBC 1–2, WBC 1–2, hyaline cylinders: none, bacteria, few, squamous epithelia 10–20, dipstick analysis, blood moderate positive). This was interpreted as a urinary tract infections and treated with antibiotics. Her emergency record states that the patient developed hemoglobinuria after being treated with trimethoprim-sulfamethoxazole (co-trimoxazole). Hemogobinuria was associated with lightheadedness and dizziness as well as a mild increase of her liver enzymes (aspartate aminotransferase 77U/L (14–36)). She was therefore screened twice for Glucose-6-Phosphate-Dehydrogenase (G6DP) deficiency, but showed enzyme activity levels within normal limits. At the age of 20-years she became pregnant. She continued to have a picture of hemolytic anemia (hemoglobin range between 9–10 g/dL with a reticulocyte count of 4%, LDH of 555 U/L, total bilirubin range 0.4–0.5 mg/dL, haptoglobin of 2 mg/dL (41–165mg/dL)) but this time, she had negative cold agglutinins and negative direct Coombs test. Her pregnancy was complicated with the worsening of her anemia and development of thrombocytopenia requiring red cell and platelet transfusions. At 34 weeks of gestation she was seen by a hematologist who sent for PNH testing. Flow cytometry revealed a significant population of PNH cells in both red blood and white blood cells (65% of red blood cells and 94% of granulocytes) with a large proportion of red cells with an intermediate expression of CD59 (58%, granulocytes 12%). She was started on anticoagulation with low molecular heparin. She had intermittent episodes of overt hemoglobinuria. At 35 weeks of gestation she was diagnosed with severe hypertension and was admitted for the induction of labor. Her hospital course was complicated with the development of preeclampsia and an acute flair of hemolysis leading to acute renal failure requiring hemodialysis. A healthy baby was delivered by cesarean section. She was vaccinated against meningococcal infection and initiated on eculizumab (Soliris, Alexion Pharmaceuticals) with prophylactic antibiotics for the first 14 days. Her anticoagulation was switched from heparin to warfarin. Her renal functions recovered to normal over the next three months. The patient decided to discontinue eculizumab and warfarin on her own as she thought that this was not beneficial and she continued to have episodes of dark urine.
Three years later, she presented again with left upper abdominal pain, vomiting and blurry vision. She was diagnosed with an acute hemolytic exacerbation of PNH and was admitted. The patient was found to have anemia, leukocytosis and mild transaminitis. A Magnetic Resonace Venogram (MRV) of the abdomen and pelvis was obtained and it showed non-specific perfusional abnormalities throughout the liver. A Computed Tomography (CT) of the abdomen and pelvis revealed several ill-defined low attenuation lesions of the posterior segment of the right hepatic lobe. This was thought to be consistent with liver thrombosis so she was restarted on anticoagulation with warfarin and was referred to us for further evaluation and treatment recommendations and to rule out resistance to C5 inhibitor therapy.
On review of systems, the patient complained of occasional abdominal pain and headaches. On physical exam, she was found to have mildly icteric sclera. Family history was significant for a sister with sickle cell trait. Further questioning revealed that that one of her nephews was diagnosed with G6PD deficiency. The laboratory workup revealed a white blood cell count 4.6 k/uL (4.5–11.0 k/uL), RBC count of 3.8 k/uL (4.2–5.5 k/uL), hemoglobin of 11.8g/dL, hematocrit 35% (37–47%), MCV 91 fl (82–100 fl), platelets 327 k/uL (150–400 k/uL), reticulocytes 24.5%, PTT 27.8 sec (23–36 sec), INR 1.7, D dimer 2.5 ug/mL (0–0.4), bilirubin 1.3 mg/dL, bilirubin direct 0.2 mg/dL (0–0.4 mg/dL), bilirubin indirect 1.1 mg/dL, ANC 2.7 k/uL (1.75–7.59 k/uL), ALC 1.3 k/uL (1.12–4.95 k/uL), G6PD screen normal, LDH 720 U/L. The flow cytometry at this point revealed that 78% of her red blood cells were PNH, 69% were partially deficient for CD59 and 9.1% lacked CD59 completely; 92% of her granulocytes were PNH, 86% were partially deficient for CD59 and 6% lacked the expression of CD59. Due to her history of hepatic vein thrombosis eculizumab was reinitiated at its regular dosing for patients with PNH (600 mg weekly 4x followed by 900 mg every two weeks). In three years of being on eculizumab she had no further relapse of her hemoglobinuria and no evidence of thrombosis.
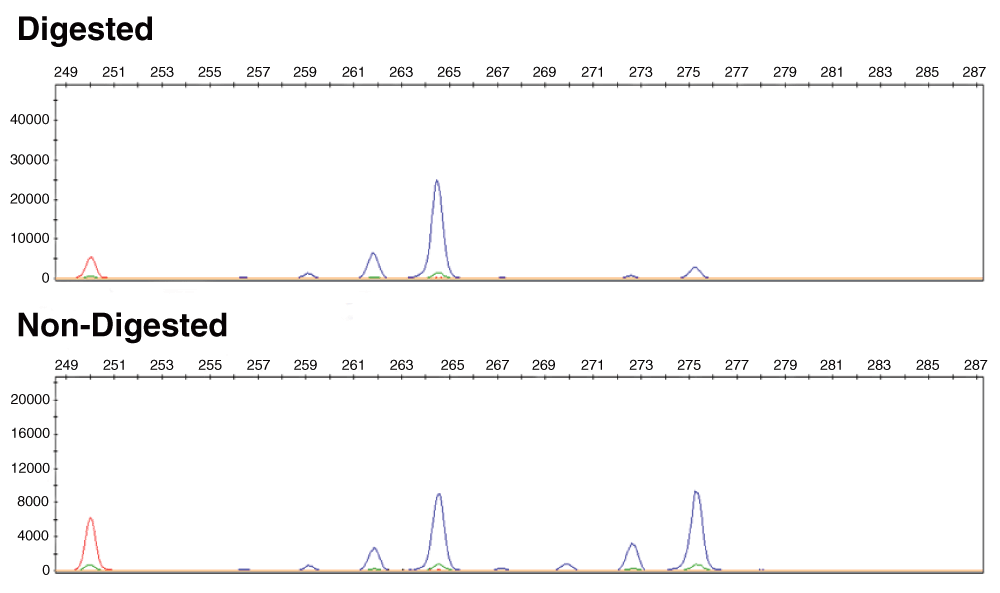
Results
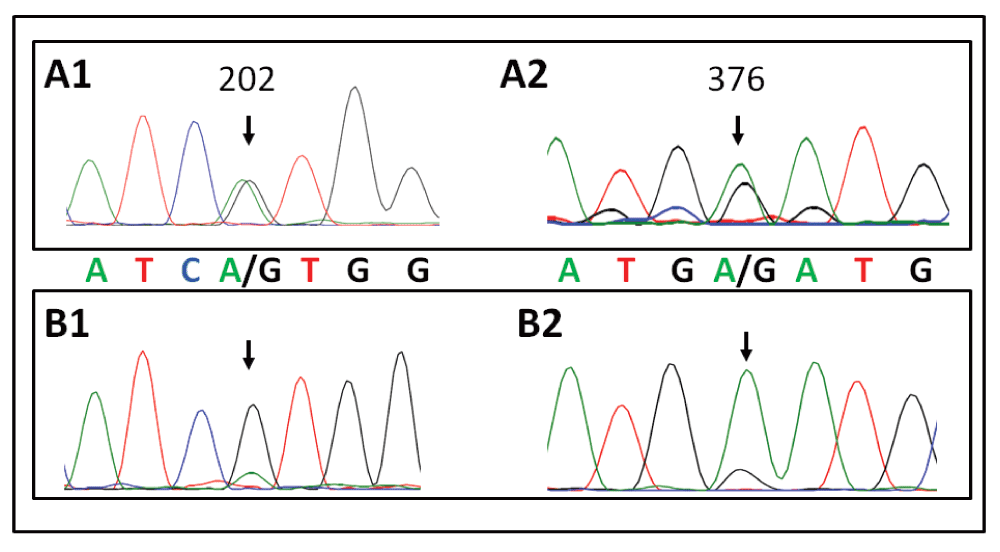
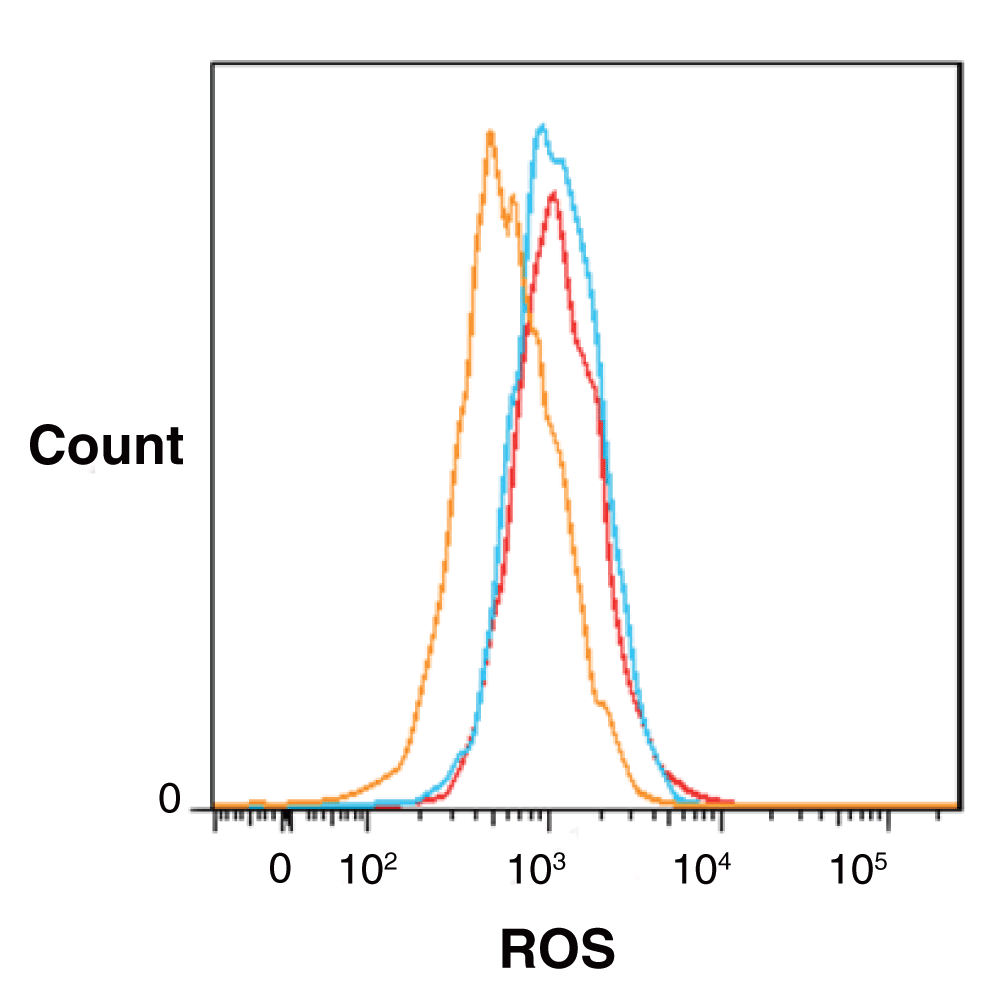
This patient has a classic presentation of a patient whose blood cells mainly have a partial deficiency of GPI-linked proteins (PNH type II) with a significant delay in diagnosis relative infrequent hemolytic events and thrombotic complications. We were intrigued by the emergency physicians note that associated hemoglobinuria and clinical symptoms associated with hemolysis with the co-trimoxazole medication and family history of G6PD deficiency. We were therefore interested to find out whether she actually might have both, and whether her PNH might have been responsible for her G6PD deficiency and thereby explain the hemolysis precipitated by co-trimoxazole as noted by an obervant emergency physician. Sequencing of DNA from her granulocytes confirmed that she was heterozygous for G6PD A- having a G6PD B allele on one X-chromosome and a G6PD A- allele on the other. This finding raised the question as to whether the somatic PIGA mutation causing her PNH took place on the X-chromosome carrying the B or the A- G6PD gene. The flow cytometry data showed that the patient most likely had 2 PNH clones, a class II clone (partial deficiency) of about 86% and a class III clone (complete deficiency) of about 6%. The HUMARA assay, which measures X-inactivation, showed a single clone of about 90% (Figure 1), suggesting that in both clones the mutation had taken place on the same X-chromosome. We hypothesized that the mutations in PIGA would have taken place on the chromosome carrying the G6PD A- allele since this would help explain the patient’s reaction to co-trimoxazole. To determine which G6PD allele was expressed in the PNH clone we sequenced cDNA from the patient’s granulocytes. The sequencing trace showed that the vast majority of expressed G6PD cDNA contained the wild type (G6PD B) sequence at both nucleotides where it differs from G6PD A- (Figure 2), leaving us to conclude that the PIGA mutations had taken place on the X-chromosome containing the G6PD B allele. This was confirmed at the protein level since the red blood cells lysed by acidified serum (the PNH cells) contained most of the G6PD activity while the residual cells did not contain detectable G6PD activity. While developing our hypothesis, which turned out to be incorrect, we also considered whether PNH/G6PDA- cells might have high levels of oxidative stress since both G6PD deficiency and PNH have been shown to be associated with elevated levels of reactive oxygen species11,13. We found that the patient’s red blood cells contained ROS levels that were significantly higher than those from healthy controls, though surprisingly we did not detect any difference in ROS between PNH (CD55-) and normal (CD55+) cells (Figure 3).
Figure 1. Clonal hematopoiesis in patient CHOP 277.01.
The panel at the bottom shows the migration of the 2 microsatellite alleles in patient CHOP277.01 revealed by PCR analysis of a region of the Androgen receptor gene on the X-chromosome. The amplified region contains both a polymorphic repeat and a site for the methylation sensitive restriction enzyme HhaI. This site is methylated on the inactive X-chromosome. The top panel shows the same sample digested with HhaI before the PCR so only the fragment on the inactive X-chromosome is amplified. An imbalance in the allelic ratio reflects an imbalance in X inactivation and therefore indicates clonality.
Figure 2. Patient CHOP 277.01. is heterozygous for G6PD B/A- and the B allele is predominantly expressed.
A1 and A2 shows sequencing of genomic DNA around the 202 G->A and the 376 A->G mutation that lead to Val68Met and Asn126Asp changes in G6PD, respectively. Figures B1 and B2 show the cDNA sequence of the exact same mutations in the peripheral blood of the patient. The WT (B) allele is expressed in the majority of cells.
Figure 3. Flow cytometry analysis of the oxidative status of RBCs in the patient and a healthy donor.
Peripheral blood mononuclear cells from the patient and a normal control were treated with the oxidation sensitive dye, CM-H2DCFDA, and the conversion to its oxidized fluorescent derivative assessed by flow cytometry. The fluorescence distribution histogram and the mean fluorescence channels (MFC) of each population derived from the normal control (orange) and the patient (blue for CD55 negative and red for CD55 positive cells) are shown. The figure shows a representative example of 4 normal controls that gave similar results.
Discussion
PNH is a rare condition, having an incidence of about 1 in a million, so the co-incidental finding of a female with PNH and heterozygous for G6PD deficiency was an opportunity to observe the interaction between these 2 conditions which both involve red blood cell hemolysis mediated by X-linked genes. Notably the first demonstration that PNH was a clonal disease took advantage of a female PNH patient who was heterozygous for the electrophoretic variant G6PD A14. In this patient both isozymes were present in a lysate of total red blood cells, but only one was present after acidified serum lysis, demonstrating clonality of the PNH cells.
In the case discussed here a female African American patient with PNH suffered episodes of hemolysis, often following treatment with Trimethoprim-Sulfamethoxazole (co-trimoxazole), one of the drugs that is known to cause hemolysis in G6PD patients15. When it emerged that she was heterozygous for G6PD A- we hypothesized that her expanded PNH clones may be expressing only the G6PD A- protein, which would have explained the observation of the emergency physician. The hypothesis proved incorrect and the clone expressed the wild type G6PD allele. An alternative intriguing explanation for the co-trimoxazole associated hemolysis might be that complement activation on G6PD A- red cells exposed to co-trimoxazole might have triggered complement activation inducing the lysis of G6PD B PNH Type II red blood cells16. naturally we cannot rule out that at the time the co-trimoxazole associated hemolysis was observed a G6PD A- PNH clone was more prevalent, since PNH patients, like the one described here, often have several PNH clones17. Of course it is also possible that co-trimoxazole medication and hemolysis were coincidental and that hemolysis was primarily due to the urinary tract infection. Nevertheless the observation by an unbiased emergency physician and a rather bland urine sediment make us favor the first explanation. Finally, the association of PNH and G6PD deficiency make us also speculate that the combination of G6PD and PIGA deficiency confers a serious growth disadvantage and PNH clones in this situation are more likely to be G6PD wild type. There is no clear mechanism for this however as G6PDA- nucleated cells have similar enzyme activity to WT cells18 – the deficiency becoming apparent in red blood cells, which do not synthesize new protein19.
Patient consent
Written informed consent for publication of their clinical details was obtained from the patient.
Author contributions
PJM and MB conceived the study, NP designed the experiments and carried out the research. MM collected and collated clinical data. All authors contributed to preparing a draft of the manuscript and have agreed to the final content.
Competing interests
No competing interests were disclosed
Grant information
The work has been supported by the Buck Family Endowed Chair in Hematology, and by NCI NIH grants 2R01CA106995 to PJ Mason, and 2R01 CA105312 to M Bessler.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
2.
Luzzatto L, Bessler M, Rotoli B:
Somatic mutations in paroxysmal nocturnal hemoglobinuria: a blessing in disguise?
Cell.
1997; 88(1): 1–4. PubMed Abstract
| Publisher Full Text
3.
McKeage K:
Eculizumab: a review of its use in paroxysmal nocturnal haemoglobinuria.
Drugs.
2011; 71(17): 2327–45. PubMed Abstract
| Publisher Full Text
4.
Kelly RJ, Hill A, Arnold LM, et al.:
Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival.
Blood.
2011; 117(25): 6786–92. PubMed Abstract
| Publisher Full Text
5.
Mason PJ, Bautista JM, Gilsanz F:
G6PD deficiency: the genotype-phenotype association.
Blood Rev.
2007; 21(5): 267–83. PubMed Abstract
| Publisher Full Text
8.
Hirono A, Beutler E:
Molecular cloning and nucleotide sequence of cDNA for human glucose-6-phosphate dehydrogenase variant A(-).
Proc Natl Acad Sci U S A.
1988; 85(11): 3951–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
9.
Au WY, Lam V, Pang A, et al.:
Glucose-6-phosphate dehydrogenase deficiency in female octogenarians, nanogenarians, and centenarians.
J Gerontol A Biol Sci Med Sci.
2006; 61(10): 1086–9. PubMed Abstract
| Publisher Full Text
10.
Allen RC, Zoghbi HY, Moseley AB, et al.:
Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation.
Am J Hum Genet.
1992; 51(6): 1229–39. PubMed Abstract
| Free Full Text
11.
Amer J, Zelig O, Fibach E:
Oxidative status of red blood cells, neutrophils, and platelets in paroxysmal nocturnal hemoglobinuria.
Exp Hematol.
2008; 36(4): 369–77. PubMed Abstract
| Publisher Full Text
12.
Rattazzi MC, Corash LM, van Zanen GE, et al.:
G6PD deficiency and chronic hemolysis: four new mutants--relationships between clinical syndrome and enzyme kinetics.
Blood.
1971; 38(2): 205–18. PubMed Abstract
13.
Ronquist G, Theodorsson E:
Inherited, non-spherocytic haemolysis due to deficiency of glucose-6-phosphate dehydrogenase.
Scand J Clin Lab Invest.
2007; 67(1): 105–11. PubMed Abstract
| Publisher Full Text
14.
Oni SB, Osunkoya BO, Luzzatto L:
Paroxysmal nocturnal hemoglobinuria: evidence for monoclonal origin of abnormal red cells.
Blood.
1970; 36(2): 145–52. PubMed Abstract
15.
Serpa JA, Villarreal-Williams E, Giordano TP:
Prevalence of G6PD deficiency in a large cohort of HIV-infected patients.
J Infect.
2010; 61(5): 399–402. PubMed Abstract
| Publisher Full Text
16.
Arese P, Gallo V, Pantaleo A, et al.:
Life and Death of Glucose-6-Phosphate Dehydrogenase (G6PD) Deficient Erythrocytes - Role of Redox Stress and Band 3 Modifications.
Transfus Med Hemother.
2012; 39(5): 328–334. PubMed Abstract
| Publisher Full Text
| Free Full Text
17.
Mortazavi Y, Merk B, McIntosh J, et al.:
The spectrum of PIG-A gene mutations in aplastic anemia/paroxysmal nocturnal hemoglobinuria (AA/PNH): a high incidence of multiple mutations and evidence of a mutational hot spot.
Blood.
2003; 101(7): 2833–41. PubMed Abstract
| Publisher Full Text
18.
Marks PA, Gross RT:
Erythrocyte glucose-6-phosphate dehydrogenase deficiency: evidence of differences between Negroes and Caucasians with respect to this genetically determined trait.
J Clin Invest.
1959; 38(12): 2253–62. PubMed Abstract
| Publisher Full Text
| Free Full Text
19.
Mason PJ:
New insights into G6PD deficiency.
Br J Haematol.
1996; 94(4): 585–91. PubMed Abstract
1
Division of Hematology, Department of Pediatrics, Abramson Research Center, The Children’s Hospital of Philadelphia, Philadelphia, 19104, USA 2
Division of Hematology, University of Pennsylvania School of Medicine, Philadelphia, 19104-4318, USA
The work has been supported by the Buck Family Endowed Chair in Hematology, and by NCI NIH grants 2R01CA106995 to PJ Mason, and 2R01 CA105312 to M Bessler.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Perdigones N, Morales M, Mason P and Bessler M. Case Report: Paroxysmal nocturnal hemoglobinuria in a woman heterozygous for G6PD A- [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2014, 3:194 (https://doi.org/10.12688/f1000research.4980.2)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
NOTE: it is important to ensure the information in square brackets after the title is included in this citation.
Reviewer Report18 Feb 2015
José M. Bautista,
Department of Biochemistry and Molecular Biology IV and Research Institute Hospital 12 de Octubre, Complutense University of Madrid, Madrid, Spain
This case report describes the rare concurrence of two haematological genetic disorders associated to chromosome X (PNH and G6PD deficiency) and provides a retrospective interpretation of the clinical development throughout the detailed and focused experimental analysis of the patient’s cells
... Continue reading
This case report describes the rare concurrence of two haematological genetic disorders associated to chromosome X (PNH and G6PD deficiency) and provides a retrospective interpretation of the clinical development throughout the detailed and focused experimental analysis of the patient’s cells and the genetics associated to them. Following an appealing and reasonable hypothesis, the results obtained by Perdigones et al. offer an authoritative haematology lesson. Thus, the authors take the case report as a centre to discuss the biology of PNH clones and the potential mechanism of red cell lysis when two independent haemolysis-prone genes are associated in the same patient.
The originality and presentation of the case warrant the readers interest. The final version with the amendments made following the report from two previous reviewers resulted in a solid article worth reading for haematologists and geneticists.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Perdigones and colleagues describe the rare co-occurence in the same patient of two X-linked disorders, one acquired the other inherited, both causing intravascular haemolysis.
However how the interaction of the two disorders led to the clinical episode described in the case
... Continue reading
Perdigones and colleagues describe the rare co-occurence in the same patient of two X-linked disorders, one acquired the other inherited, both causing intravascular haemolysis.
However how the interaction of the two disorders led to the clinical episode described in the case report is not clear because the temporal analysis of the clinical events and the associated laboratory tests are not presented in sufficient detail.
Charting clinical events, labs and therapeutic interventions might make association of the heamolytic episodes with co-trimoxazole or intercurrent infection clearer.
It appears that the majority of the PNH clone (in both granulocytes and red cells) is type II, i.e., only partially deficient of GPI. One assumes that this picture was obtained after the haemolytic attack. If so, it could be that type III, i.e., severely deficient RBC were indeed G6PD-deficient. Was flow-cytometry performed after Eculizumab treatment? Eculizumab would protect type III RBC from lysis and thus would allow re-assessment of G6PD activity.
Minor points
Was anti-CD55 or –CD59 was used for flow analysis? The authors state anti-CD55 in methods but describe anti-CD59 in results.
Normal ranges of lab tests need to be provided
‘Another factor was that treatment with eculizumab, by inhibiting lysis of PNH red cells may have led to a higher level of PNH (and concomitantly G6PD deficient) red cells than would be present in untreated patients.’ This statement is rather irrelevant because eculizumab was started after treatment with co-trimoxazole.
Need to use Greek characters appropriately, i.e., ul should be ml
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however I have significant reservations, as outlined above.
Author Response
(F1000Research Advisory Board Member)
15 Oct 2014
Philip Mason, The Children's Hospital of Philadelphia, USA
15 Oct 2014
Author Response F1000Research Advisory Board Member
It appears that the majority of the PNH clone (in both granulocytes and red cells) is type II, i.e., only partially deficient of GPI. One assumes that this picture was
...
Continue readingIt appears that the majority of the PNH clone (in both granulocytes and red cells) is type II, i.e., only partially deficient of GPI. One assumes that this picture was obtained after the haemolytic attack. If so, it could be that type III, i.e., severely deficient RBC were indeed G6PD-deficient. Was flow-cytometry performed after Eculizumab treatment? Eculizumab would protect type III RBC from lysis and thus would allow re-assessment of G6PD activity.
We agree with this comment. Of course it is possible that at the time of the observation of association of co-trimoxazole associated hemolysis a different PNH clone was more prevalent than when analyzed for G6PD deficiency. We included this possibility in our discussion. The most recent flow cytometry was performed when the patient was on eculizimab.
Minor points
Was anti-CD55 or –CD59 was used for flow analysis? The authors state anti-CD55 in methods but describe anti-CD59 in results. The diagnosis for PNH was performed in a CLIA approved clinical laboratory of the hospital.
We present the results for CD59. CD55 was also tested however the discrimination between the three populations is more difficult.
Normal ranges of lab tests need to be provided.
We have included normal ranges for laboratory tests in parentheses after the patient’s values.
Another factor was that treatment with eculizumab, by inhibiting lysis of PNH red cells may have led to a higher level of PNH (and concomitantly G6PD deficient) red cells than would be present in untreated patients.’ This statement is rather irrelevant because eculizumab was started after treatment with co-trimoxazole.
We think this statement is valid because treatment with eculizimab increases the level of PIGA deficient red cells. On our hypothesis that the G6PDA- allele was linked with PIGA- then PIGA-,G6PDA- red cells would increase. Our hypothesis however was not correct.
Need to use Greek characters appropriately, i.e., ul should be ml
We have corrected our incorrect use of Greek characters.
It appears that the majority of the PNH clone (in both granulocytes and red cells) is type II, i.e., only partially deficient of GPI. One assumes that this picture was obtained after the haemolytic attack. If so, it could be that type III, i.e., severely deficient RBC were indeed G6PD-deficient. Was flow-cytometry performed after Eculizumab treatment? Eculizumab would protect type III RBC from lysis and thus would allow re-assessment of G6PD activity.
We agree with this comment. Of course it is possible that at the time of the observation of association of co-trimoxazole associated hemolysis a different PNH clone was more prevalent than when analyzed for G6PD deficiency. We included this possibility in our discussion. The most recent flow cytometry was performed when the patient was on eculizimab.
Minor points
Was anti-CD55 or –CD59 was used for flow analysis? The authors state anti-CD55 in methods but describe anti-CD59 in results. The diagnosis for PNH was performed in a CLIA approved clinical laboratory of the hospital.
We present the results for CD59. CD55 was also tested however the discrimination between the three populations is more difficult.
Normal ranges of lab tests need to be provided.
We have included normal ranges for laboratory tests in parentheses after the patient’s values.
Another factor was that treatment with eculizumab, by inhibiting lysis of PNH red cells may have led to a higher level of PNH (and concomitantly G6PD deficient) red cells than would be present in untreated patients.’ This statement is rather irrelevant because eculizumab was started after treatment with co-trimoxazole.
We think this statement is valid because treatment with eculizimab increases the level of PIGA deficient red cells. On our hypothesis that the G6PDA- allele was linked with PIGA- then PIGA-,G6PDA- red cells would increase. Our hypothesis however was not correct.
Need to use Greek characters appropriately, i.e., ul should be ml
We have corrected our incorrect use of Greek characters.
Competing Interests:No competing interests were disclosed.Close
Author Response
(F1000Research Advisory Board Member)
15 Oct 2014
Philip Mason, The Children's Hospital of Philadelphia, USA
15 Oct 2014
Author Response F1000Research Advisory Board Member
It appears that the majority of the PNH clone (in both granulocytes and red cells) is type II, i.e., only partially deficient of GPI. One assumes that this picture was
...
Continue readingIt appears that the majority of the PNH clone (in both granulocytes and red cells) is type II, i.e., only partially deficient of GPI. One assumes that this picture was obtained after the haemolytic attack. If so, it could be that type III, i.e., severely deficient RBC were indeed G6PD-deficient. Was flow-cytometry performed after Eculizumab treatment? Eculizumab would protect type III RBC from lysis and thus would allow re-assessment of G6PD activity.
We agree with this comment. Of course it is possible that at the time of the observation of association of co-trimoxazole associated hemolysis a different PNH clone was more prevalent than when analyzed for G6PD deficiency. We included this possibility in our discussion. The most recent flow cytometry was performed when the patient was on eculizimab.
Minor points
Was anti-CD55 or –CD59 was used for flow analysis? The authors state anti-CD55 in methods but describe anti-CD59 in results. The diagnosis for PNH was performed in a CLIA approved clinical laboratory of the hospital.
We present the results for CD59. CD55 was also tested however the discrimination between the three populations is more difficult.
Normal ranges of lab tests need to be provided.
We have included normal ranges for laboratory tests in parentheses after the patient’s values.
Another factor was that treatment with eculizumab, by inhibiting lysis of PNH red cells may have led to a higher level of PNH (and concomitantly G6PD deficient) red cells than would be present in untreated patients.’ This statement is rather irrelevant because eculizumab was started after treatment with co-trimoxazole.
We think this statement is valid because treatment with eculizimab increases the level of PIGA deficient red cells. On our hypothesis that the G6PDA- allele was linked with PIGA- then PIGA-,G6PDA- red cells would increase. Our hypothesis however was not correct.
Need to use Greek characters appropriately, i.e., ul should be ml
We have corrected our incorrect use of Greek characters.
It appears that the majority of the PNH clone (in both granulocytes and red cells) is type II, i.e., only partially deficient of GPI. One assumes that this picture was obtained after the haemolytic attack. If so, it could be that type III, i.e., severely deficient RBC were indeed G6PD-deficient. Was flow-cytometry performed after Eculizumab treatment? Eculizumab would protect type III RBC from lysis and thus would allow re-assessment of G6PD activity.
We agree with this comment. Of course it is possible that at the time of the observation of association of co-trimoxazole associated hemolysis a different PNH clone was more prevalent than when analyzed for G6PD deficiency. We included this possibility in our discussion. The most recent flow cytometry was performed when the patient was on eculizimab.
Minor points
Was anti-CD55 or –CD59 was used for flow analysis? The authors state anti-CD55 in methods but describe anti-CD59 in results. The diagnosis for PNH was performed in a CLIA approved clinical laboratory of the hospital.
We present the results for CD59. CD55 was also tested however the discrimination between the three populations is more difficult.
Normal ranges of lab tests need to be provided.
We have included normal ranges for laboratory tests in parentheses after the patient’s values.
Another factor was that treatment with eculizumab, by inhibiting lysis of PNH red cells may have led to a higher level of PNH (and concomitantly G6PD deficient) red cells than would be present in untreated patients.’ This statement is rather irrelevant because eculizumab was started after treatment with co-trimoxazole.
We think this statement is valid because treatment with eculizimab increases the level of PIGA deficient red cells. On our hypothesis that the G6PDA- allele was linked with PIGA- then PIGA-,G6PDA- red cells would increase. Our hypothesis however was not correct.
Need to use Greek characters appropriately, i.e., ul should be ml
We have corrected our incorrect use of Greek characters.
Competing Interests:No competing interests were disclosed.Close
Perdigones and collaborators report an interesting clinical case about the association of PNH and G6PD deficiency. However, the timing of the clinical history is not clear and the report is scanty of some relevant clinical/laboratory data.
Major scientific points.
The Authors should
... Continue reading
Perdigones and collaborators report an interesting clinical case about the association of PNH and G6PD deficiency. However, the timing of the clinical history is not clear and the report is scanty of some relevant clinical/laboratory data.
Major scientific points.
The Authors should provide the exact timing of entire clinical history: diagnosis; infective episode resulting in the prescription of trimethoprim-sulfamethoxazole -co-trimoxazole- (which dose?); start and stop of co-trimoxazole; “dark urine episode” associated with co-trimoxazole; start of eculizumab; time of biological studies.
It is extremely important that the Authors provide more details about the timing and the features of the hemolytic attack apparently associated with co-trimoxazole: how long after the start of co-trimoxazole treatment the patient experienced the “dark urine episode”? There are any objective data at the time of this “dark urine episode” or the episode has been just self-reported? At the time of this “dark urine episode” there were any signs/symptoms of an ongoing infective condition?
The Authors provide clinical/laboratory details only at one time point, that seems be after the start of eculizumab (how long after?). They should provide such clinical/laboratory details at time of diagnosis, at the time of the co-trimoxazole associated “hemolytic attack”, at start of eculizumab treatment, etc.: blood count, absolute reticulocyte count, LDH levels (providing the normal range), flow citometry, etc.
In PNH patients the presence of red blood cells with partial deficiency of GPI-linked molecules is relatively common. However, the presence of granulocyte/monocyte with partial deficiency of GPI-linked molecules is uncommon: thus, it would be interesting to show the dot plot of the “CD59/lineage marker” analysis of granulocytes and monocytes of this patient.
The Authors, at variance with their starting hypothesis, have clearly proven that in this patient PNH cells express the wild type G6PD B. They provide 2 possible explanations for the co-trimoxazole-associated “hemolytic crisis” observed in this patient. These hypotheses are interesting but their probability is very low. I suggest that the Authors should discuss a much more likely explanation: this “hemolytic crisis” was just due to the infective condition that led to the prescription of co-trimoxazole.
Minor points
In the Introduction the Authors report the classical classification of PNH cells as Type III (completely deficient in GPI anchored proteins), Type II (partially deficient) and Type I (normal display of GPI-linked proteins). However, in the Results they write about “class I clone (partial deficiency) … and class II clone (complete deficiency) “: this is extremely confusing.
In the Results the 2 sentences (from “We hypothesized that …” to “…red cells than would be present in untreated patients.”) seem to suggest that the relative expansion of the “PNH (and concomitantly G6PD deficient) red cells” following eculizumab treatment could have played a role in the co-trimoxazole associated hemolytic attack. The Authors should rephrase these sentences since eculizumab has been started after the co-trimoxazole associated hemolytic attack.
Trimethoprim-sulfamethoxazole (co-trimoxazole) is a generic drug name thus it should not be capitalized. In the Results, “eculizumab” should not be capitalized.
The brand name Bactrim should be replaced with the generic drug name.
The levels of ROS in the patient red cells should be compared with a group of healthy controls: the comparison with only one control does not allow any conclusion.
The Authors should report the concentration of DCF used for ROS detection.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however I have significant reservations, as outlined above.
Author Response
(F1000Research Advisory Board Member)
15 Oct 2014
Philip Mason, The Children's Hospital of Philadelphia, USA
15 Oct 2014
Author Response F1000Research Advisory Board Member
Perdigones and collaborators report an interesting clinical case about the association of PNH and G6PD deficiency. However, the timing of the clinical history is not clear and the report is
...
Continue readingPerdigones and collaborators report an interesting clinical case about the association of PNH and G6PD deficiency. However, the timing of the clinical history is not clear and the report is scanty of some relevant clinical/laboratory data.
Major scientific points.
The Authors should provide the exact timing of entire clinical history: diagnosis; infective episode resulting in the prescription of trimethoprim-sulfamethoxazole -co-trimoxazole- (which dose?); start and stop of co-trimoxazole; “dark urine episode” associated with co-trimoxazole; start of eculizumab; time of biological studies.
In our revised manuscript we include a fuller, more detailed case history. Of note is the fact that the patient presented to the specialty clinic with a probably 6 year history of PNH and that most of the patients past history relies on records and notes. Here we demonstrate that the patient has indeed both conditions PNH and G6PD deficiency. With these results in hand we try to explain the patient’s history and clinical observations. It was not our intention to prove that indeed the hemolysis described by the emergency physician after co-trimoxazole was indeed triggered by the drug, but rather to evaluate and discuss the possibility as the patient carries on her chart the diagnosis of co-trimoxazole hypersensitity due to this incident. We try to make this clearer in the case description. We hope this will answer the criticism of the reviewer.
It is extremely important that the Authors provide more details about the timing and the features of the hemolytic attack apparently associated with co-trimoxazole: how long after the start of co-trimoxazoletreatment the patient experienced the “dark urine episode”? There are any objective data at the time of this “dark urine episode” or the episode has been just self-reported? At the time of this “dark urine episode” there were any signs/symptoms of an ongoing infective condition?
The discussion above and the revised version provides many more details.
The Authors provide clinical/laboratory details only at one time point, that seems be after the start of eculizumab (how long after?). They should provide such clinical/laboratory details at time of diagnosis, at the time of the co-trimoxazole associated “hemolytic attack”, at start of eculizumab treatment, etc.: blood count, absolute reticulocyte count, LDH levels (providing the normal range), flow citometry, etc.
We have provided all the history we can obtain in the revised version.
In PNH patients the presence of red blood cells with partial deficiency of GPI-linked molecules is relatively common. However, the presence of granulocyte/monocyte with partial deficiency of GPI-linked molecules is uncommon: thus, it would be interesting to show the dot plot of the “CD59/lineage marker” analysis of granulocytes and monocytes of this patient.
The diagnosis of PNH and the subtype analysis of PNH granulocytes was performed in a CLIA approved clinical laboratory, and their test results are reported here. The individual dot blots are not available to us. However the senior author who was involved in the setup and quality assessment of flow-cytometric PNH testing at this institution fully trusts their analysis. We agree with the reviewer that partial GPI-anchor deficiency in granulocytes is less frequently observed and very much depends on the underlying mutation and the antibody chosen for the analysis.
The Authors, at variance with their starting hypothesis, have clearly proven that in this patient PNH cells express the wild type G6PD B. They provide 2 possible explanations for the co-trimoxazole-associated “hemolytic crisis” observed in this patient. These hypotheses are interesting but their probability is very low. I suggest that the Authors should discuss a much more likely explanation: this “hemolytic crisis” was just due to the infective condition that led to the prescription of co-trimoxazole.
We agree that this is a possible alternative explanation and have added this in the discussion however considering the rather bland urine sediment we actually favor the other two possible explanations.
Minor points
In the Introduction the Authors report the classical classification of PNH cells as Type III (completely deficient in GPI anchored proteins), Type II (partially deficient) and Type I (normal display of GPI-linked proteins). However, in the Results they write about “class I clone (partial deficiency) … and class II clone (complete deficiency) “: this is extremely confusing.
This has been corrected
In the Results the 2 sentences (from “We hypothesized that …” to “…red cells than would be present in untreated patients.”) seem to suggest that the relative expansion of the “PNH (and concomitantly G6PD deficient) red cells” following eculizumab treatment could have played a role in the co-trimoxazole associated hemolytic attack. The Authors should rephrase these sentences since eculizumab has been started after the co-trimoxazole associated hemolytic attack.
The timing of the eculizimab treatment and the hemolysis are given in the detailed case report that we now provide.
Trimethoprim-sulfamethoxazole (co-trimoxazole) is a generic drug name thus it should not be capitalized. In the Results, “eculizumab” should not be capitalized.
We have corrected this.
The brand name Bactrim should be replaced with the generic drug name.
We have corrected this.
The levels of ROS in the patient red cells should be compared with a group of healthy controls: the comparison with only one control does not allow any conclusion.
The experiment was carried out with several controls and it confirms the results of Amer et al., cited as our ref 14 that ROS are elevated in PNH. Our point is a small one, that we might expect a further elevation if we compare PNH and normal cells from this patient, if indeed PIGA- is linked with G6PDA-. We didn’t find this. We include the sentence “The figure shows a representative example of 4 normal controls that gave similar results” to clarify this.
The Authors should report the concentration of DCF used for ROS detection.
We have done this.
Perdigones and collaborators report an interesting clinical case about the association of PNH and G6PD deficiency. However, the timing of the clinical history is not clear and the report is scanty of some relevant clinical/laboratory data.
Major scientific points.
The Authors should provide the exact timing of entire clinical history: diagnosis; infective episode resulting in the prescription of trimethoprim-sulfamethoxazole -co-trimoxazole- (which dose?); start and stop of co-trimoxazole; “dark urine episode” associated with co-trimoxazole; start of eculizumab; time of biological studies.
In our revised manuscript we include a fuller, more detailed case history. Of note is the fact that the patient presented to the specialty clinic with a probably 6 year history of PNH and that most of the patients past history relies on records and notes. Here we demonstrate that the patient has indeed both conditions PNH and G6PD deficiency. With these results in hand we try to explain the patient’s history and clinical observations. It was not our intention to prove that indeed the hemolysis described by the emergency physician after co-trimoxazole was indeed triggered by the drug, but rather to evaluate and discuss the possibility as the patient carries on her chart the diagnosis of co-trimoxazole hypersensitity due to this incident. We try to make this clearer in the case description. We hope this will answer the criticism of the reviewer.
It is extremely important that the Authors provide more details about the timing and the features of the hemolytic attack apparently associated with co-trimoxazole: how long after the start of co-trimoxazoletreatment the patient experienced the “dark urine episode”? There are any objective data at the time of this “dark urine episode” or the episode has been just self-reported? At the time of this “dark urine episode” there were any signs/symptoms of an ongoing infective condition?
The discussion above and the revised version provides many more details.
The Authors provide clinical/laboratory details only at one time point, that seems be after the start of eculizumab (how long after?). They should provide such clinical/laboratory details at time of diagnosis, at the time of the co-trimoxazole associated “hemolytic attack”, at start of eculizumab treatment, etc.: blood count, absolute reticulocyte count, LDH levels (providing the normal range), flow citometry, etc.
We have provided all the history we can obtain in the revised version.
In PNH patients the presence of red blood cells with partial deficiency of GPI-linked molecules is relatively common. However, the presence of granulocyte/monocyte with partial deficiency of GPI-linked molecules is uncommon: thus, it would be interesting to show the dot plot of the “CD59/lineage marker” analysis of granulocytes and monocytes of this patient.
The diagnosis of PNH and the subtype analysis of PNH granulocytes was performed in a CLIA approved clinical laboratory, and their test results are reported here. The individual dot blots are not available to us. However the senior author who was involved in the setup and quality assessment of flow-cytometric PNH testing at this institution fully trusts their analysis. We agree with the reviewer that partial GPI-anchor deficiency in granulocytes is less frequently observed and very much depends on the underlying mutation and the antibody chosen for the analysis.
The Authors, at variance with their starting hypothesis, have clearly proven that in this patient PNH cells express the wild type G6PD B. They provide 2 possible explanations for the co-trimoxazole-associated “hemolytic crisis” observed in this patient. These hypotheses are interesting but their probability is very low. I suggest that the Authors should discuss a much more likely explanation: this “hemolytic crisis” was just due to the infective condition that led to the prescription of co-trimoxazole.
We agree that this is a possible alternative explanation and have added this in the discussion however considering the rather bland urine sediment we actually favor the other two possible explanations.
Minor points
In the Introduction the Authors report the classical classification of PNH cells as Type III (completely deficient in GPI anchored proteins), Type II (partially deficient) and Type I (normal display of GPI-linked proteins). However, in the Results they write about “class I clone (partial deficiency) … and class II clone (complete deficiency) “: this is extremely confusing.
This has been corrected
In the Results the 2 sentences (from “We hypothesized that …” to “…red cells than would be present in untreated patients.”) seem to suggest that the relative expansion of the “PNH (and concomitantly G6PD deficient) red cells” following eculizumab treatment could have played a role in the co-trimoxazole associated hemolytic attack. The Authors should rephrase these sentences since eculizumab has been started after the co-trimoxazole associated hemolytic attack.
The timing of the eculizimab treatment and the hemolysis are given in the detailed case report that we now provide.
Trimethoprim-sulfamethoxazole (co-trimoxazole) is a generic drug name thus it should not be capitalized. In the Results, “eculizumab” should not be capitalized.
We have corrected this.
The brand name Bactrim should be replaced with the generic drug name.
We have corrected this.
The levels of ROS in the patient red cells should be compared with a group of healthy controls: the comparison with only one control does not allow any conclusion.
The experiment was carried out with several controls and it confirms the results of Amer et al., cited as our ref 14 that ROS are elevated in PNH. Our point is a small one, that we might expect a further elevation if we compare PNH and normal cells from this patient, if indeed PIGA- is linked with G6PDA-. We didn’t find this. We include the sentence “The figure shows a representative example of 4 normal controls that gave similar results” to clarify this.
The Authors should report the concentration of DCF used for ROS detection.
We have done this.
Competing Interests:No competing interests were disclosed.Close
Author Response
(F1000Research Advisory Board Member)
15 Oct 2014
Philip Mason, The Children's Hospital of Philadelphia, USA
15 Oct 2014
Author Response F1000Research Advisory Board Member
Perdigones and collaborators report an interesting clinical case about the association of PNH and G6PD deficiency. However, the timing of the clinical history is not clear and the report is
...
Continue readingPerdigones and collaborators report an interesting clinical case about the association of PNH and G6PD deficiency. However, the timing of the clinical history is not clear and the report is scanty of some relevant clinical/laboratory data.
Major scientific points.
The Authors should provide the exact timing of entire clinical history: diagnosis; infective episode resulting in the prescription of trimethoprim-sulfamethoxazole -co-trimoxazole- (which dose?); start and stop of co-trimoxazole; “dark urine episode” associated with co-trimoxazole; start of eculizumab; time of biological studies.
In our revised manuscript we include a fuller, more detailed case history. Of note is the fact that the patient presented to the specialty clinic with a probably 6 year history of PNH and that most of the patients past history relies on records and notes. Here we demonstrate that the patient has indeed both conditions PNH and G6PD deficiency. With these results in hand we try to explain the patient’s history and clinical observations. It was not our intention to prove that indeed the hemolysis described by the emergency physician after co-trimoxazole was indeed triggered by the drug, but rather to evaluate and discuss the possibility as the patient carries on her chart the diagnosis of co-trimoxazole hypersensitity due to this incident. We try to make this clearer in the case description. We hope this will answer the criticism of the reviewer.
It is extremely important that the Authors provide more details about the timing and the features of the hemolytic attack apparently associated with co-trimoxazole: how long after the start of co-trimoxazoletreatment the patient experienced the “dark urine episode”? There are any objective data at the time of this “dark urine episode” or the episode has been just self-reported? At the time of this “dark urine episode” there were any signs/symptoms of an ongoing infective condition?
The discussion above and the revised version provides many more details.
The Authors provide clinical/laboratory details only at one time point, that seems be after the start of eculizumab (how long after?). They should provide such clinical/laboratory details at time of diagnosis, at the time of the co-trimoxazole associated “hemolytic attack”, at start of eculizumab treatment, etc.: blood count, absolute reticulocyte count, LDH levels (providing the normal range), flow citometry, etc.
We have provided all the history we can obtain in the revised version.
In PNH patients the presence of red blood cells with partial deficiency of GPI-linked molecules is relatively common. However, the presence of granulocyte/monocyte with partial deficiency of GPI-linked molecules is uncommon: thus, it would be interesting to show the dot plot of the “CD59/lineage marker” analysis of granulocytes and monocytes of this patient.
The diagnosis of PNH and the subtype analysis of PNH granulocytes was performed in a CLIA approved clinical laboratory, and their test results are reported here. The individual dot blots are not available to us. However the senior author who was involved in the setup and quality assessment of flow-cytometric PNH testing at this institution fully trusts their analysis. We agree with the reviewer that partial GPI-anchor deficiency in granulocytes is less frequently observed and very much depends on the underlying mutation and the antibody chosen for the analysis.
The Authors, at variance with their starting hypothesis, have clearly proven that in this patient PNH cells express the wild type G6PD B. They provide 2 possible explanations for the co-trimoxazole-associated “hemolytic crisis” observed in this patient. These hypotheses are interesting but their probability is very low. I suggest that the Authors should discuss a much more likely explanation: this “hemolytic crisis” was just due to the infective condition that led to the prescription of co-trimoxazole.
We agree that this is a possible alternative explanation and have added this in the discussion however considering the rather bland urine sediment we actually favor the other two possible explanations.
Minor points
In the Introduction the Authors report the classical classification of PNH cells as Type III (completely deficient in GPI anchored proteins), Type II (partially deficient) and Type I (normal display of GPI-linked proteins). However, in the Results they write about “class I clone (partial deficiency) … and class II clone (complete deficiency) “: this is extremely confusing.
This has been corrected
In the Results the 2 sentences (from “We hypothesized that …” to “…red cells than would be present in untreated patients.”) seem to suggest that the relative expansion of the “PNH (and concomitantly G6PD deficient) red cells” following eculizumab treatment could have played a role in the co-trimoxazole associated hemolytic attack. The Authors should rephrase these sentences since eculizumab has been started after the co-trimoxazole associated hemolytic attack.
The timing of the eculizimab treatment and the hemolysis are given in the detailed case report that we now provide.
Trimethoprim-sulfamethoxazole (co-trimoxazole) is a generic drug name thus it should not be capitalized. In the Results, “eculizumab” should not be capitalized.
We have corrected this.
The brand name Bactrim should be replaced with the generic drug name.
We have corrected this.
The levels of ROS in the patient red cells should be compared with a group of healthy controls: the comparison with only one control does not allow any conclusion.
The experiment was carried out with several controls and it confirms the results of Amer et al., cited as our ref 14 that ROS are elevated in PNH. Our point is a small one, that we might expect a further elevation if we compare PNH and normal cells from this patient, if indeed PIGA- is linked with G6PDA-. We didn’t find this. We include the sentence “The figure shows a representative example of 4 normal controls that gave similar results” to clarify this.
The Authors should report the concentration of DCF used for ROS detection.
We have done this.
Perdigones and collaborators report an interesting clinical case about the association of PNH and G6PD deficiency. However, the timing of the clinical history is not clear and the report is scanty of some relevant clinical/laboratory data.
Major scientific points.
The Authors should provide the exact timing of entire clinical history: diagnosis; infective episode resulting in the prescription of trimethoprim-sulfamethoxazole -co-trimoxazole- (which dose?); start and stop of co-trimoxazole; “dark urine episode” associated with co-trimoxazole; start of eculizumab; time of biological studies.
In our revised manuscript we include a fuller, more detailed case history. Of note is the fact that the patient presented to the specialty clinic with a probably 6 year history of PNH and that most of the patients past history relies on records and notes. Here we demonstrate that the patient has indeed both conditions PNH and G6PD deficiency. With these results in hand we try to explain the patient’s history and clinical observations. It was not our intention to prove that indeed the hemolysis described by the emergency physician after co-trimoxazole was indeed triggered by the drug, but rather to evaluate and discuss the possibility as the patient carries on her chart the diagnosis of co-trimoxazole hypersensitity due to this incident. We try to make this clearer in the case description. We hope this will answer the criticism of the reviewer.
It is extremely important that the Authors provide more details about the timing and the features of the hemolytic attack apparently associated with co-trimoxazole: how long after the start of co-trimoxazoletreatment the patient experienced the “dark urine episode”? There are any objective data at the time of this “dark urine episode” or the episode has been just self-reported? At the time of this “dark urine episode” there were any signs/symptoms of an ongoing infective condition?
The discussion above and the revised version provides many more details.
The Authors provide clinical/laboratory details only at one time point, that seems be after the start of eculizumab (how long after?). They should provide such clinical/laboratory details at time of diagnosis, at the time of the co-trimoxazole associated “hemolytic attack”, at start of eculizumab treatment, etc.: blood count, absolute reticulocyte count, LDH levels (providing the normal range), flow citometry, etc.
We have provided all the history we can obtain in the revised version.
In PNH patients the presence of red blood cells with partial deficiency of GPI-linked molecules is relatively common. However, the presence of granulocyte/monocyte with partial deficiency of GPI-linked molecules is uncommon: thus, it would be interesting to show the dot plot of the “CD59/lineage marker” analysis of granulocytes and monocytes of this patient.
The diagnosis of PNH and the subtype analysis of PNH granulocytes was performed in a CLIA approved clinical laboratory, and their test results are reported here. The individual dot blots are not available to us. However the senior author who was involved in the setup and quality assessment of flow-cytometric PNH testing at this institution fully trusts their analysis. We agree with the reviewer that partial GPI-anchor deficiency in granulocytes is less frequently observed and very much depends on the underlying mutation and the antibody chosen for the analysis.
The Authors, at variance with their starting hypothesis, have clearly proven that in this patient PNH cells express the wild type G6PD B. They provide 2 possible explanations for the co-trimoxazole-associated “hemolytic crisis” observed in this patient. These hypotheses are interesting but their probability is very low. I suggest that the Authors should discuss a much more likely explanation: this “hemolytic crisis” was just due to the infective condition that led to the prescription of co-trimoxazole.
We agree that this is a possible alternative explanation and have added this in the discussion however considering the rather bland urine sediment we actually favor the other two possible explanations.
Minor points
In the Introduction the Authors report the classical classification of PNH cells as Type III (completely deficient in GPI anchored proteins), Type II (partially deficient) and Type I (normal display of GPI-linked proteins). However, in the Results they write about “class I clone (partial deficiency) … and class II clone (complete deficiency) “: this is extremely confusing.
This has been corrected
In the Results the 2 sentences (from “We hypothesized that …” to “…red cells than would be present in untreated patients.”) seem to suggest that the relative expansion of the “PNH (and concomitantly G6PD deficient) red cells” following eculizumab treatment could have played a role in the co-trimoxazole associated hemolytic attack. The Authors should rephrase these sentences since eculizumab has been started after the co-trimoxazole associated hemolytic attack.
The timing of the eculizimab treatment and the hemolysis are given in the detailed case report that we now provide.
Trimethoprim-sulfamethoxazole (co-trimoxazole) is a generic drug name thus it should not be capitalized. In the Results, “eculizumab” should not be capitalized.
We have corrected this.
The brand name Bactrim should be replaced with the generic drug name.
We have corrected this.
The levels of ROS in the patient red cells should be compared with a group of healthy controls: the comparison with only one control does not allow any conclusion.
The experiment was carried out with several controls and it confirms the results of Amer et al., cited as our ref 14 that ROS are elevated in PNH. Our point is a small one, that we might expect a further elevation if we compare PNH and normal cells from this patient, if indeed PIGA- is linked with G6PDA-. We didn’t find this. We include the sentence “The figure shows a representative example of 4 normal controls that gave similar results” to clarify this.
The Authors should report the concentration of DCF used for ROS detection.
We have done this.
Competing Interests:No competing interests were disclosed.Close
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)