Introduction
A protein structure is formed by well ordered local segments defined by the hydrogen-bonding pattern of the peptide backbone (secondary structures), and conformations that lack any regular arrangement (random coils). The most prevalent secondary structures are alpha helices (AH) and β sheets, while other conformations like π-helix occur rarely in natural proteins1. AHs are right-handed spiral conformations which have a hydrogen bond between the carbonyl oxygen (C=O) of every residue and the alpha-amino nitrogen (N-H) of the fourth residue away from the N-terminal.
DSSP is the official program used to assign secondary structure to a protein when the atomic coordinates are known2,3. Several methods can also predict an AH from the sequence4,5. Essentially, any structure prediction tool can be used to predict an AH from the sequence by first predicting the structure and then applying DSSP to the predicted structure6–8.
The niche of AHs in protein structures is widespread. AHs are the functionally significant element in several motifs (DNA binding motifs)9, and the key components of any protein that permeates biological membranes10. AHs are also almost invariably present in anti-microbial peptides (AMP)11. For example, cecropin B, a component of a chimeric protein with anti-microbial properties that provides grapevines with enhanced resistance against the Gram-negative pathogen Xylella fastidiosa12, is composed of two AHs connected by a small random coil13. Other AMPs comprise only a single AH14,15. These peptides are characterized by a strong hydrophobic surface (defined by a hydrophobic moment16), and often have charged residues, either anionic or cationic, aligned on the opposite surface16. Previously, Jones et al. have implemented computational methods to extract the characteristics of AHs17.
In the current work, we first observe and propose an empirical structural property of AHs: that the distance between the Cα atoms of the ith and (i+4)th residue is equal to the distance between the carbonyl oxygens of the ith and (i+4)th residue. This hypothesis is validated on a set of high resolution non-homologous 100 proteins (775 AHs) taken from the PISCES database18. Next, we implement the methodologies described previously17 to compute the hydrophobic moments for AHs using the hydrophobicity scale used in19: PAGAL - Properties and corresponding graphics of alpha helical structures in proteins. There are other programs available online to do similar processing (http://rzlab.ucr.edu/scripts/wheel/ for example). We also specify a metric associated with each helix - the ratio of the positive to the negative residues (RPNR) in the AH - which helps identify AHs with a particular kind of charge distribution on their surface. The results are outputted as the input to a graphical program TikZ (for the Edmundson wheel20 and hydrophobic moment), and Pymol scripts (for showing the peptide surface). The source code and manual available at http://github.com/sanchak/pagal and on http://dx.doi.org/10.5281/zenodo.11136.
Materials and methods
We first outline the method to obtain the coordinates of each residue in the Edmundson wheel, and the computation of the hydrophobic moment (Algorithm 1). The input to the function is an alpha helix - either as a PDB structure or as a fasta sequence. The center of the wheel is taken as (0,0) and the radius as 5. The first residue has coordinates (0,5). Each subsequent residue is advanced by 100 degrees on the circle, as 3.6 turns of the helix makes one full circle.
To compute the hydrophobic moment, we obtain the vector by connecting the center to the coordinate of the residue and giving it a magnitude obtained from the hydrophobic scale (in our case, this scale is obtained from17). These vectors are then added to obtain the final hydrophobic moment.
The results are outputted as the input to a graphical program TiKz (for the Edmundson wheel20 and hydrophobic moment), and Pymol scripts (for showing the peptide surface). The protein structures have been rendered using Pymol, while the figures showing the Edmundson wheel has been obtained from TiKz. The source code is written in Perl, and made available at https://github.com/sanchak/pagal and permanently available on http://dx.doi.org/10.5281/zenodo.11136.
Algorithm 1. Calculate hydrophobic moment
Input: αH: α helix - either PDB or fasta sequence
Input: TableHS: Hydrophobic scale
Output: TikZIN: TikZ input file
Output: PymolIN: Pymol input file
begin
Radius = 5 ; // Radius of Edmundson wheel
initangle = 90 ; // first residue is at 12 o’clock..
loopcnt = 0 ;
finalvechydro = undefined ;
centre = (0,0);
foreach Residuei in αH do
/* Find X,Y coordinate on the Edmundson wheel */
angle = initangle - loopcnt * 100 ;
x = Radius * cos(val) ;
y = Radius * sin(val) ;
thispoint = (x,y);
/* Get Hydrophobic moment */
vector = MakeVectorFrom2Points(centre,thispoint) ;
hydrophobicvalue = GetHydrophobicScaleForResidue(TableHS, Residuei) ;
tmpvec = normal(vector) * hydrophobicvalue ;
finalvechydro = finalvechydro is not defined? tmpvec : finalvechydro + tmpvec;
loopcnt++ ;
end
WriteTikzScript();
WritePymolScript();
end
Results and discussion
Validation of empirical property
We have observed an empirical structural property that applies to the residues of any AH: the distance between the Cα atoms of the ith and (i+4)th residue (denoted by D(Cαi/Cαi+4)) is (almost) equal to the distance between the carbonyl oxygens of the ith and (i+4)th residue (D(Oi/Oi+4)). We validate our hypothesis on a set of 100 high resolution, non-homologous proteins (which have 775 AHs) taken from the PISCES database (http://dunbrack.fccc.edu/PISCES.php)18. Figure 1 shows the plot of the difference between D(Cαi/Cαi+4) and D(Oi/Oi+4) for AHs specified in the PDB files (in red, mean=0.16 Å, standard deviation (sd)=0.34 Å), and for all residues separated by four residues but not part of a helix (in blue, mean=0.71 Å, sd=0.75 Å).
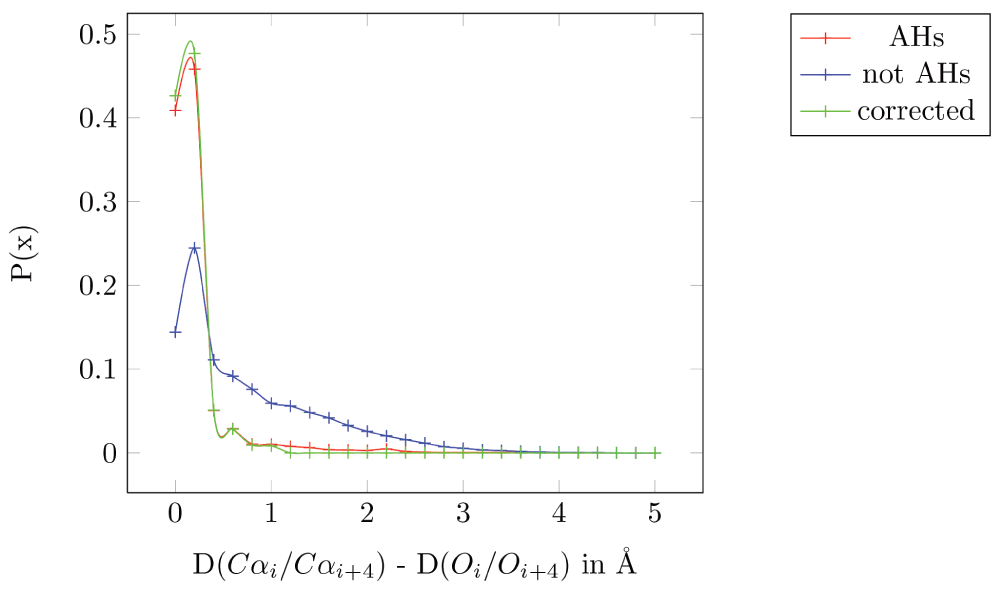
Figure 1. Plot of the difference between D(Cαi/Cαi+4) and D(Oi/Oi+4).
All 775 AHs specified in the PDB files from the 100 non-homologous high resolution structures taken from the PISCES database are in red (mean=0.16 Å, standard deviation (sd)=0.34α Å). All residues separated by four residues but not part of a helix are in blue (mean=0.71 Å, sd=0.75 Å). All AHs specified in the PDB files after correction are in green (mean=0.095 Å and sd=0.14 Å).
These results are conservative, since there are residues that are annotated as part of a helix in the PDB file which seems to be incorrect. For example, in PBD 1JET, the ninth helix spans from residues 169 to 178 - “HELIX 9 9 LYS A 169 LYS A 178 1 10”. However, the Pymol helix identification program shows part of this stretch as a random coil (Lys178 in Figure 2a). Moreover, the distance between the carbonyl oxygen (C=O) and the alpha-amino nitrogen (N-H) of the fourth residue away from the N-terminal is 7.6 Å, which makes it improbable for them to have a hydrogen bond, the primary requisite to be part of a AH. The D(Cαi/Cαi+4) and D(Oi/Oi+4) for this pair is 9 Å and 8 Å, respectively: a difference of 1 Å. Even in cases where the distance between C=O and N-H is within the 3.6 Å typically required for a hydrogen bond, (PDBid: 1ELU, 12th helix), the distances D(Cαi/Cαi+4) and D(Oi/Oi+4) for the residue pair His292-Gly296 is 6.9 Å and 3.4 Å, respectively: a difference of 3.4 Å (Figure 2b). In short, the helix annotation in the PDB database is often incorrect. Removing these problematic residues reduces the mean distance to 0.095 Å and the sd to 0.14 Å (Figure 1).

Figure 2. Incorrect annotations of helices in the PDB file.
(a) Lys178 in PDBid:1JET appears to be part of a random coil, but is annotated in the PDB file as a helix. (b) Gly296 in PDBid:1ELU is mis-annotated similarly.
There is variation in the D(Cαi/Cαi+4) even when considering the same pair of residues. For example, taking all pairs of Arg and Lys in the 775 AHs analyzed (Table 1), we see that the values can vary from 6.5 Å in PDBid:1H16 (helix26, pair Arg583-Lys587) to 5.8 Å in PDBid:1EYH (helix5, pair Arg72-Lys76). However, as hypothesized, D(Oi/Oi+4) is the same as D(Cαi/Cαi+4).
Table 1. Occurences of Arg-Lys pairs in 775 alpha helices found in 100 non-homologous high resolution protein structures taken from the PISCES database:
RPair: Residue pair in the alpha helix with a hydrogen bond between carbonyl oxygen (C=O) and the alpha-amino nitrogen (N-H), Dhbond: Distance between carbonyl oxygen (C=O) and the alpha-amino nitrogen (N-H) of RPair, D(Cαi/Cαi+4): Distance between the Cα atoms of RPair, D(Oi/Oi+4): Distance between the carbonyl oxygen of RPair, δ: absolute(D(Cαi/Cαi+4) - D(Oi/Oi+4)).
| Helix | Residue Pair | Dhbond | D(Cαi/Cαi+4) | D(Oi/Oi+4) | δ |
|---|
| 1E58.helix12 | Arg188-Lys192 | 2.9 | 6.0 | 6.1 | 0.1 |
| 1H16.helix26 | Arg583-Lys587 | 3.3 | 6.5 | 6.5 | 0.0 |
| 1ELK.helix4 | Arg52-Lys56 | 2.9 | 6.1 | 6.2 | 0.1 |
| 1EYH.helix5 | Arg72-Lys76 | 2.9 | 5.8 | 5.8 | 0.0 |
| 1F1E.helix4 | Arg89-Lys93 | 2.9 | 6.1 | 6.1 | 0.0 |
| 1GXM.helix9 | Arg481-Lys485 | 2.7 | 5.9 | 6.0 | 0.1 |
| 1JET.helix14 | Arg290-Lys294 | 3.0 | 6.3 | 6.2 | 0.1 |
| 1EYH.helix9 | Arg124-Lys128 | 3.0 | 6.2 | 6.2 | 0.0 |
| 1GCI.helix7 | Arg247-Lys251 | 3.1 | 6.4 | 6.3 | 0.1 |
| 1EB6.helix3 | Arg60-Lys64 | 3.1 | 6.4 | 6.4 | 0.0 |
| 1DK8.helix3 | Arg140-Lys144 | 2.9 | 6.1 | 6.1 | 0.0 |
| 1GKP.helix5 | Arg192-Lys196 | 2.9 | 6.1 | 6.1 | 0.0 |
| 1D5T.helix8 | Arg138-Lys142 | 3.1 | 6.4 | 6.4 | 0.0 |
Edmundson wheel and the hydrophobic moment
The Edmundson wheel20 has been the standard way of visualizing AHs for a long time now, although there are other methods (Wenxiang diagram21) to represent AHs. The Edmundson wheel shows the alignment of residues as one looks through the helix, and gives an approximate idea of the various properties of the AH. For example, a color coding differentiation of the polar and non-polar residues gives an approximation of the hydrophobic propensity of the AH. A more mathematical representation of the hydrophobic propensity is to represent each residue with a value and a sign (direction). This results in a vector representation, called the hydrophobic moment16. We have chosen the hydrophobic scale from17 (Table 2), although any other hydrophobic scale could be also used. The color coding is as follows: all hydrophobic residues (positive values in Table 2) are colored red, while hydrophilic residues (negative values in Table 2) are colored in blue: dark blue for positively charged residues, medium blue for negatively charged residues and light blue for amides. We now show the PAGAL representation of a few AH peptides.
Table 2. Hydrophobicity scale taken from17.
MET
0.975 | ILE
0.913 | LEU
0.852 | VAL
0.811 | CYS
0.689 | ALA
0.607 | THR
0.525 | GLY
0.484 | SER
0.402 | HIS
0.333 |
PRO
0.239 | PHE
1.036 | TRP
0.668 | TYR
0.137 | GLN
-0.558 | ASN
-0.701 | GLU
-1.396 | LYS
-1.518 | ASP
-1.600 | ARG
-2.233 |
Cecropin. A synergistic combination of two critical immune functions, pathogen surface recognition and lysis, resulted in a chimeric protein with anti-microbial properties against the Gram-negative Xylella fastidiosa12. The lytic domain is cecropin B, which attacks conserved lipid moieties and creates pores in the X. fastidiosa outer membrane13. Cecropin B consists of two AHs, joined by a short stretch of random coil. Figure 3a and b shows the Edmundson wheel and hydrophobic moment of the two AHs. It can be seen that the N-Terminal AH has a large hydrophobic moment, as well as a specific positive charge distribution. The hydrophobicity of this amphipathic AH has significant bearing on the anti-microbial properties of the peptide22. This can also be seen in a Pymol rendering of the peptide surface (Figure 4). The Pymol script for this rendering is automatically generated by PAGAL. On the other hand, the C-Terminal AH comprises mostly of hydrophobic residues. Cecropin-like peptides use the synergy of these two helices - the N-terminal attaches to charged ion on the membrane, and the hydrophobic C-terminal permeates the hydrophobic inter-membrane region (known as the ‘carpet’ model23).
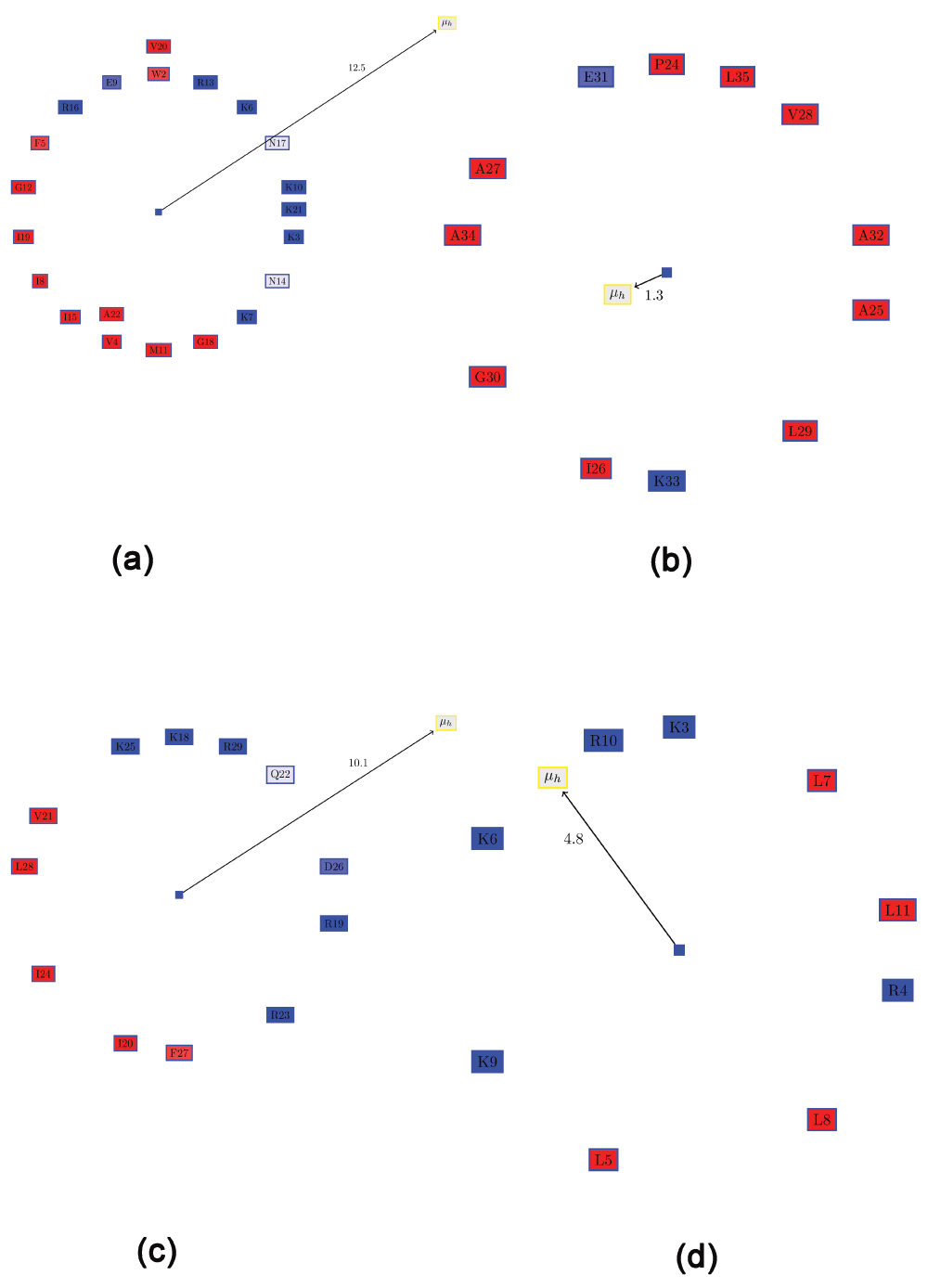
Figure 3. Visualizing the Edmundson wheel and hydrophobic moment of some alpha helices.
All hydrophobic residues are colored in red, while hydrophilic residues are colored in blue: dark blue for positively charged residues, medium blue for negatively charged residues and light blue for amides. (a) N-Terminal helix of cecropin B. (b) C-Terminal helix of cecropin B. (c) KR-12 peptide fragment from cathelicidin LL-37. (d) De novo designed peptide (SP1-1) with anti-microbial activity.
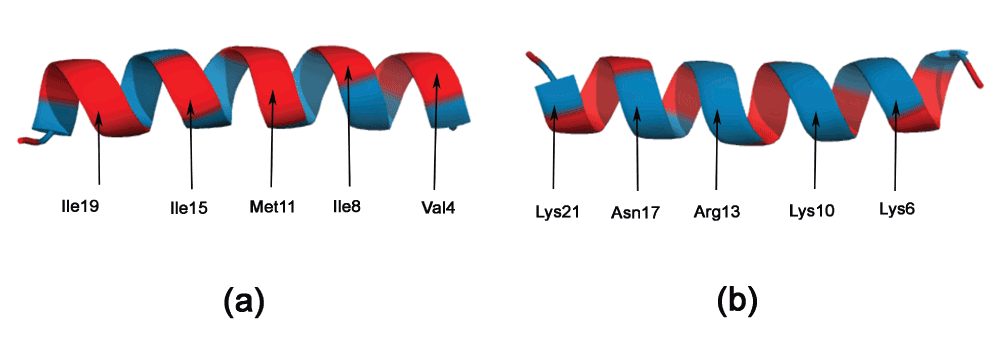
Figure 4. Pymol rendering of peptides showing the hydrophobic and charged surfaces for the N-terminal helix of cecropin B.
All hydrophobic residues are colored in red, while hydrophilic residues are colored in blue.
Cathelicidin LL-37. Cathelicidin LL-37 is a critical component of the innate human immune system that protects humans against infectious diseases by targeting anionic phosphatidylglycerols in the pathogenic bacterial membranes24. Recent work has demonstrated a 12-residue peptide (KR-12) corresponding to residues 18 to 29 of LL-37 is toxic to bacterial, but not human cells14. Figure 3c shows the Edmundson wheel and hydrophobic moment of KR-12. The demarcation of the polar and non-polar residues is quite evident. The predominance of positively charged residues in the polar side of the peptide is also clearly visible.
De novo designed AMPs for plant protection. The de novo design of small AMPs that inhibit plant pathogens was the focus of a recent work15. One of the most promising candidates was a small peptide (SP1-1 - RKKRLKLLKRL, Figure 3d), which was “highly active against a broad spectrum of bacteria, but showed low hemolytic activity”15. Although the hydrophobic moment of this peptide is much smaller than that of KR-12 (Figure 3c), possibly due to the presence of Arg4 on the hydrophobic surface, the distribution of positively charged residues in this peptide is greater than for KR-12.
Ratio of the positive to the negative residues (RPNR)
Often, it is desirable to choose a large distribution of charged residues of a certain kind (anionic or cationic) on the hydrophilic surface. One possible method for quantifying this would be to compute a ‘charge moment’, similar to the computation of hydrophobic moments. However, such an evaluation would determine certain clearly distributions to be the same. For example, assume one semicircle of the wheel comprised only positive residues, and the other hydrophobic residues (Figure 5a). This is a slightly modified version of KR-12 from cathelicidin LL-37. If one positive residue (R5) were moved from the hydrophilic side to the hydrophobic side (I7) and replaced with a negative residue (D7) (Figure 5b), the ‘charge moment’ would remain the same, although the two conformations are clearly not the same. Note that the hydrophobic moment is also different, as expected. Thus, we resort to a simple metric to allow one to choose peptides with a large proportion of charged residue of a single kind: the ratio of the positive to the negative residues (RPNR). The two peptides mentioned above will have different RPNRs: 1 (Figure 5a) and 0.85 (Figure 5b).
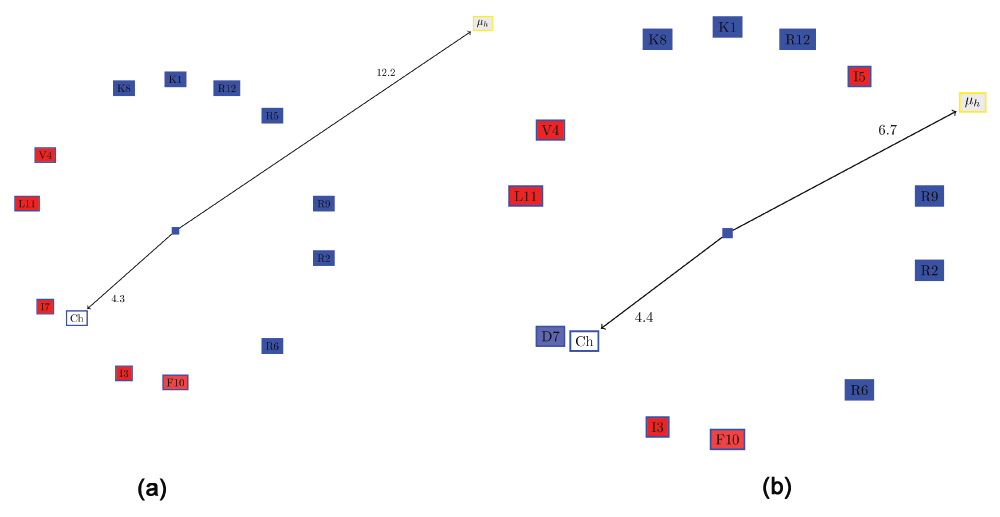
Figure 5. The problem in evaluating a ‘charge moment’ similar to the way the hydrophobic moment is computed.
All hydrophobic residues are colored in red, while the hydrophilic residues are colored in blue: dark blue for positively charged residues, medium blue for negatively charged residues and light blue for amides. (a) Edmundson wheel of a KR-12 like peptide showing the hydrophobic moment and the ‘charge moment’. (b) Swapping one positive residue (R5) from the hydrophilic side with I7 and replacing it with a negative residue (D7), results in the same ‘charge moment’, although the characteristics of the helix has clearly changed.
Output formats
PAGAL generates a TikZ input file for drawing the Edmundson wheel and showing the hydrophobic moment (Supplementary File TikzInput.doc). TikZ is a package “for creating graphics programmatically” - http://www.texample.net/tikz/. PAGAL also generates a Pymol script to the peptide structure using the same color coding used in for the Edmundson wheel (Supplementary File PymolInput.doc).
Author contributions
SC wrote the computer programs. All authors analyzed the data, and contributed equally to the writing and subsequent refinement of the manuscript.
Competing interests
No competing interests were disclosed.
Grant information
AMD wishes to acknowledge grant support from the California Department of Food and Agriculture PD/GWSS Board. BJ acknowledges financial support from Tata Institute of Fundamental Research (Department of Atomic Energy). Additionally, BJR is thankful to the Department of Science and Technology for the JC Bose Award Grant.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Faculty Opinions recommendedReferences
- 1.
Pauling L, Corey RB, Branson HR:
The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain.
Proc Natl Acad Sci U S A.
1951; 37(4): 205–211. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 2.
Kabsch W, Sander C:
Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features.
Biopolymers.
1983; 22(12): 2577–2637. PubMed Abstract
| Publisher Full Text
- 3.
Joosten RP, te Beek TA, Krieger E, et al.:
A series of PDB related databases for everyday needs.
Nucleic Acids Res.
2011; 39(Database issue): D411–419. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 4.
Maupetit J, Derreumaux P, Tuffery P:
PEP-FOLD: an online resource for de novo peptide structure prediction.
Nucleic Acids Res.
2009; 37(Web Server issue): 498–503. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 5.
Kaur H, Garg A, Raghava GP:
PEPstr: a de novo method for tertiary structure prediction of small bioactive peptides.
Protein Pept Lett.
2007; 14(7): 626–631. PubMed Abstract
| Publisher Full Text
- 6.
Arnold K, Bordoli L, Kopp J, et al.:
The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling.
Bioinformatics.
2006; 22(2): 195–201. PubMed Abstract
| Publisher Full Text
- 7.
Zhang Y:
I-TASSER server for protein 3D structure prediction.
BMC Bioinformatics.
2008; 9: 40. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 8.
Rohl CA, Strauss CE, Misura KM, et al.:
Protein structure prediction using Rosetta.
Methods Enzymol.
2004; 383: 66–93. PubMed Abstract
| Publisher Full Text
- 9.
Landschulz WH, Johnson PF, McKnight SL:
The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins.
Science.
1988; 240(4860): 1759–1764. PubMed Abstract
| Publisher Full Text
- 10.
Dathe M, Wieprecht T:
Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells.
Biochim Biophys Acta.
1999; 1462(1–2): 71–87. PubMed Abstract
| Publisher Full Text
- 11.
Brogden KA:
Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria?
Nat Rev Microbiol.
2005; 3(3): 238–250. PubMed Abstract
| Publisher Full Text
- 12.
Dandekar AM, Gouran H, Ibanez AM, et al.:
An engineered innate immune defense protects grapevines from Pierce disease.
Proc Natl Acad Sci U S A.
2012; 109(10): 3721–3725. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
Moore AJ, Beazley WD, Bibby MC, et al.:
Antimicrobial activity of cecropins.
J Antimicrob Chemother.
1996; 37(6): 1077–1089. PubMed Abstract
| Publisher Full Text
- 14.
Wang G:
Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles.
J Biol Chem.
2008; 283(47): 32637–32643. PubMed Abstract
| Publisher Full Text
- 15.
Zeitler B, Herrera Diaz A, Dangel A, et al.:
De-novo design of antimicrobial peptides for plant protection.
PLoS One.
2013; 8(8): e71687. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 16.
Eisenberg D, Weiss RM, Terwilliger TC:
The helical hydrophobic moment: a measure of the amphiphilicity of a helix.
Nature.
1982; 299(5881): 371–374. PubMed Abstract
| Publisher Full Text
- 17.
Jones MK, Anantharamaiah GM, Segrest JP:
Computer programs to identify and classify amphipathic alpha helical domains.
J Lipid Res.
1992; 33(2): 287–296. PubMed Abstract
- 18.
Wang G, Dunbrack RL Jr:
PISCES: a protein sequence culling server.
Bioinformatics.
2003; 19(12): 1589–1591. PubMed Abstract
| Publisher Full Text
- 19.
Engelman DM, Steitz TA, Goldman A:
Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins.
Annu Rev Biophys Biophys Chem.
1986; 15: 321–353. PubMed Abstract
| Publisher Full Text
- 20.
Schiffer M, Edmundson AB:
Use of helical wheels to represent the structures of proteins and to identify segments with helical potential.
Biophys J.
1967; 7(2): 121–135. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 21.
Chou KC, Zhang CT, Maggiora GM:
Disposition of amphiphilic helices in heteropolar environments.
Proteins.
1997; 28(1): 99–108. PubMed Abstract
| Publisher Full Text
- 22.
Chen Y, Guarnieri MT, Vasil AI, et al.:
Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides.
Antimicrob Agents Chemother.
2007; 51(4): 1398–1406. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 23.
Steiner H, Andreu D, Merrifield RB:
Binding and action of cecropin and cecropin analogues: antibacterial peptides from insects.
Biochim Biophys Acta.
1988; 939(2): 260–266. PubMed Abstract
| Publisher Full Text
- 24.
Yang D, Chertov O, Oppenheim JJ:
Participation of mammalian defensins and cathelicidins in anti-microbial immunity: receptors and activities of human defensins and cathelicidin (LL-37).
J Leukoc Biol.
2001; 69(5): 691–697. PubMed Abstract
- 25.
Chakraborty S, Rao BJ, Dandekar AM:
PAGAL: alpha helices structures.
Zenodo.
2014. Data Source
Comments on this article Comments (0)