Introduction
The protein GLI1, originally isolated in 1987 due to high levels of amplification in malignant glioma (Kinzler et al., 1987), is a member of the GLI family of transcription factors. This family also includes GLI2 and GLI3. GLI1 is highly conserved from Drosophila to humans and is required for developmental response via transcriptional regulation of target genes (Dennler et al., 2007; Hui & Angers, 2011; Javelaud et al., 2012). The GLI proteins, including GLI1, are transcriptional mediators of Hedgehog (HH) signaling, and regulate multiple cellular processes such as cell fate determination, tissue patterning, proliferation and transformation, which give this transcription factor a significant role in carcinogenesis if deregulated (Dennler et al., 2007; Hui & Angers, 2011; Javelaud et al., 2012). GLI1 is expressed in different human malignancies including pancreatic ductal adenocarcinoma (PDAC) (Eberl et al., 2012; Fiaschi et al., 2009; Goel et al., 2013; Hui & Angers, 2011; Mills et al., 2013; Rajurkar et al., 2012; Thayer et al., 2003). In PDAC, GLI1 is prevalently expressed in the stroma, in response to HH ligands secreted by the epithelial cells (Yauch et al., 2008). However, lower epithelial expression of Gli1 has also been reported, possibly with non-canonical functions (Nolan-Stevaux et al., 2009).
GLI1 as an oncogene in PDAC
GLI1 plays a key role in PDAC initiation by modulating the activity of two different cellular compartments, the epithelium and stroma. Rajurkar et al. demonstrated that targeted overexpression of GLI1 in the pancreas epithelium accelerates PDAC initiation by KRAS, a small GTPase mutated in more than 90% of PDAC cases (Rajurkar et al., 2012). Through use of a mouse model with simultaneous activation of oncogenic KRAS and inhibition of GLI1 in the pancreas epithelium, this group also demonstrated that decreased GLI1 activity reduced the incidence of KRAS-driven PDAC precursor lesions (pancreatic intraepithelial neoplasias or PanINs) and PDAC. Similarly, Mills and colleagues using a mouse model for pancreas-specific oncogenic KRAS expression (KC mice) bred on a Gli1 null background (GKO/KC) defined a key role for GLI1 on PDAC initiation through the modulation of the activity of fibroblasts (Mills et al., 2013). The KC mice developed PanIN lesions with 100% penetrance and PDAC in advanced age, while the GKO/KC mice did not develop PDAC and had increased survival rate when compared to KC mice. Histopathological analysis of the pancreata showed KC mice developed PanIN lesions and PDAC, while 80% of GKO/KC had normal pancreata.
Analysis of the molecular mechanism underlying this phenomenon reveals that GLI1 both regulates different target genes and is modulated by different signaling pathways depending on the cellular compartment. For instance, GLI1 activity is mainly modulated by the canonical HH signaling in fibroblasts (Yauch et al., 2008). This cascade is activated by binding of the ligand to the receptor Patched (Ptch), resulting in activation of the G-coupled receptor, Smoothened (Smo) (Javelaud et al., 2012; Yauch et al., 2008). Once activated, Smo induces GLI1 activation and upregulation of its target genes (Hui & Angers, 2011; McMahon et al., 2003). The HH ligand, Sonic Hedgehog (SHH), and components of the HH signaling pathway, including Ptch and Smo, are undetectable in the normal pancreas but overexpressed in PanINs and PDAC (Thayer et al., 2003). Inhibition of the HH pathway in PDAC cell-based xenograft models through Smo inhibition has been shown to reduce GLI1 activity and tumor growth (Feldmann et al., 2007; Thayer et al., 2003; Yauch et al., 2008). In addition, genomic sequencing of human pancreatic cancer samples revealed widespread mutations consistent with activation of the Hedgehog signaling pathway (Jones et al., 2008). While the association between HH activity and pancreatic cancer has been described over a decade ago, there is still uncertainty as to the downstream effect of HH activation in this disease. Mills et al. identified the cytokine IL-6 as a HH/GLI1 target gene in pancreatic fibroblasts (Mills et al., 2013). Increased IL-6 expression in the stromal compartment induces activation of STAT3 in the neighboring cancer cells, an essential molecular event for the progression of premalignant lesions in PDAC (Figure 1).
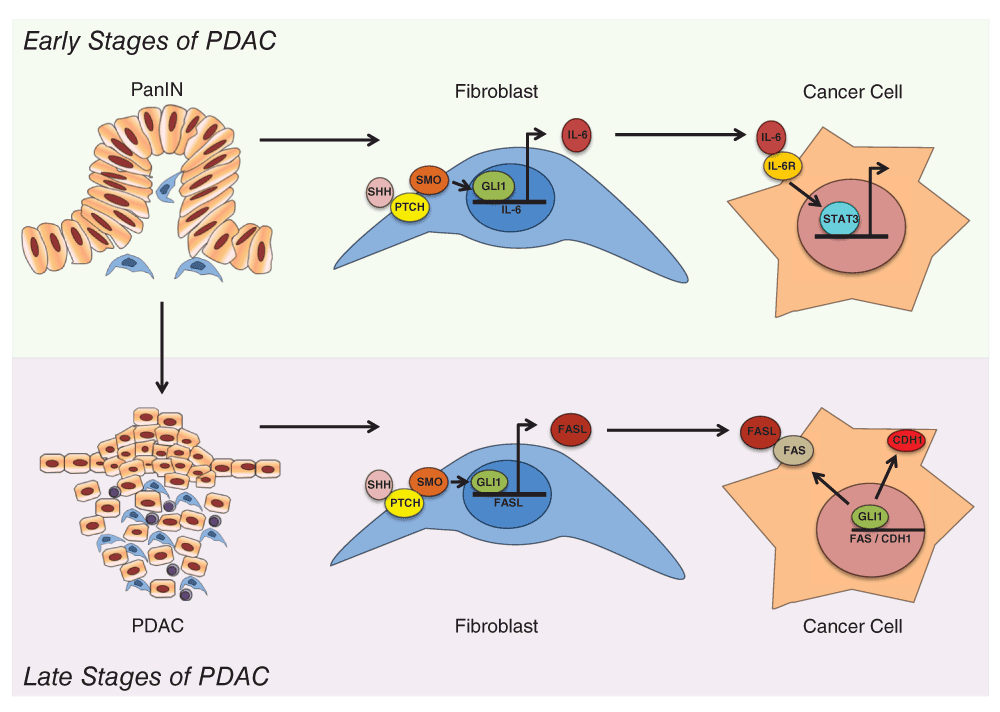
Figure 1. Working model of the dual role of GLI1 at different stages of pancreatic carcinogenesis.
During the early stages of PDAC, GLI1 is activated in the fibroblasts through canonical HH signaling. GLI1 promotes expression of the cytokine IL-6, which stimulates expression of STAT3 in neighboring cancer cells, promoting the progression of PanIN lesions to PDAC. In the later stages of PDAC, GLI1 binds the FASL promoter and regulates the expression of this ligand in the fibroblast, leading to higher levels of apoptosis in these tumors. In addition, in cancer cells, GLI1 induces the expression of FAS and CDH1 expression, leading to a tumor protective effect.
Hedgehog-independent mechanisms for GLI1 expression in PDAC
While dysregulation of HH-GLI1 signaling has been shown to play an important role in PDAC formation, several studies have demonstrated that GLI1 expression can be activated through HH-independent mechanisms in PDAC, particularly in the epithelial compartment (Dennler et al., 2007; Eberl et al., 2012; Goel et al., 2013; Ji et al., 2007; Nolan-Stevaux et al., 2009; Nye et al., 2014). Nolan-Stevaux et al. demonstrated that deletion of Smo receptor in pancreatic epithelium had no effect on KRAS induced tumor formation, nor on GLI1 expression in epithelial cells (Nolan-Stevaux et al., 2009). This indicates a Smo-independent mechanism for GLI1 regulation in PDAC cells downstream of KRAS. In fact, Ji et al. demonstrated that KRAS is a modulator of GLI1 activity and requires the transcription factor for PDAC growth in vitro (Ji et al., 2007). In the epithelial compartment, GLI1 is regulated in a HH-independent manner, downstream of KRAS. Accordingly, Ji et al. showed that Gli1 protein degradation is blocked in a MAPK-dependent manner. Furthermore, Rajurkar et al. showed a role for GLI1 in the regulation of the NF-κB pathway, a signaling cascade linked to PDAC development (Algul et al., 2002; Ougolkov et al., 2005; Pan et al., 2008; Wang et al., 1999), downstream of KRAS (Rajurkar et al., 2012). This group has identified the I-kappa-B kinase epsilon (IKBKE)/NF-κB pathway as a direct target of the GLI1 mediating KRAS-dependent pancreatic epithelial transformation in vivo (Junhao Mao, University of Massachusetts and Martin E. Fernandez-Zapico personal communication).
PDAC is characterized by a dense desmoplastic reaction associated with the primary tumor. The abundance of connective tissue is due to an increase in growth factor production in the tumor microenvironment through autocrine and paracrine oncogenic signaling pathways (Mahadevan & Von Hoff, 2007). Oncogenic KRAS activates SHH production, but HH ligands do not activate the HH pathway in tumor epithelial cells in an autocrine manner (Lauth et al., 2010; Mills et al., 2013; Yauch et al., 2008). HH signaling in PDAC occurs in a paracrine fashion where HH signaling from PDAC cells to stromal cells has been shown to promote desmoplasia (Yauch et al., 2008). Lauth et al. demonstrated that this shift from autocrine to paracrine signaling is through activation of the RAS effector dual specificity tyrosine phosphorylated and regulated kinase 1B (DYRK1B) (Lauth et al., 2010). The authors proposed this is achieved through DYRK1B inhibition of GLI2 function and promotion of the repressor GLI3, and subsequent inhibition of GLI1, in PDAC cells.
TGFβ has been shown to promote GLI1 expression in pancreatic cancer cells (Nolan-Stevaux et al., 2009). TGFβ induces the expression of GLI1 through Smad3 and LET-dependent upregulation of GLI2 independent of HH signaling (Dennler et al., 2007; Dennler et al., 2009). Nye et al. demonstrated that TGFβ, in addition to controlling GLI1 expression, can also modulate its activity by promoting the formation of a transcriptional complex with the TGFβ-regulated transcription factors, SMAD2 and 4, and the histone acetyltransferase, PCAF, in cancer cells to regulate TGFβ-induced gene expression (Nye et al., 2014). TGFβ induced GLI2 expression, and subsequent GLI1 activation, is associated with epithelial to mesenchymal transition (EMT), tumor growth, and metastasis (Javelaud et al., 2012).
In addition to TGFβ and KRAS activation, epidermal growth factor receptor (EGFR) signaling, a cascade aberrantly activated in the majority of PDACs, has been demonstrated to play a critical role in HH/GLI1-regulated tumor-initiating pancreatic cancer cells (Eberl et al., 2012). Eberl and colleagues demonstrated EGFR and HH act together to promote cancer cell proliferation by modulating gene expression through a GLI1-dependent mechanism. This suggests HH/GLI1 signaling works synergistically through distinct novel pathways during tumor initiation and growth.
Clinical trials targeting the hedgehog/GLI1 axis in PDAC
The concept that HH/GLI1 signaling might be required for PDAC growth, hence a suitable therapeutic target, has been first validated in a genetically engineered mouse model of pancreatic cancer that combines expression of oncogenic Kras with mutation of the tumor suppressor p53, the KPC mouse (Hingorani et al., 2005). Treatment of KPC mice with a Smo inhibitor in combination with gemcitabine led to a moderate but significant increase in survival (Feldmann et al., 2008; Olive et al., 2009). A preclinical study of the HH inhibitor, saridegib (IPI-926), co-administered with gemcitabine, produced a transient increase in vascular density, increased chemotherapy drug delivery, and improved disease stabilization in pancreatic cancer cells (Olive et al., 2009). Based on these results, phase II clinical trials were approved evaluating saridegib and an additional Hh inhibitor, vismodegib (GDC-0449), for treatment of pancreatic cancer. Surprisingly, the clinical trial for both vismodegib and saridegib showed a higher rate of progressive disease when compared to placebo (Catenacci et al., 2013). Similar findings were seen in a separate phase I trial of vismodegib in 8 patients with pancreatic cancer (LoRusso et al., 2011). Although hedgehog inhibitors have been successful for treating basal cell carcinoma and medulloblastoma, they do not appear to have the same effect in advanced pancreatic cancer.
These disappointing results left investigators questioning the molecular mechanism responsible for these failed clinical trials. Although there is overwhelming evidence that GLI1 plays an important role in tumor initiation and progression of several kinds of malignancies, these results suggest the transcription factor may have a tumor protective role in the later stages of certain cancers. In fact, recent studies investigating GLI1 expression in PDAC have revealed GLI1 may switch from a tumor promoting to a tumor protective molecule in the later stages of PDAC.
GLI1 as a tumor suppressor in PDAC
In contrast to the current paradigm for GLI1 expression and tumor progression, one study found GLI1 expression may actually decrease cell motility in advanced PDAC (Joost et al., 2012). Joost et al. demonstrated that GLI1 regulates epithelial differentiation through transcriptional activation of the cell adhesion molecule, E-Cadherin (CDH1), in PDAC cells. Lowered expression of GLI1 in PDAC cells lead to a loss of CDH1 expression and promotion of EMT. The transition from epithelial to motile mesenchymal cells is thought to be a critical event for metastasis of carcinomas. Decreased expression of CDH1 is associated with increased metastasis and invasion, while increased expression is associated with lower tumor malignancy (Seidel et al., 2004; von Burstin et al., 2009). PDAC is strongly associated with early invasion and metastasis. Loss of GLI1 was also shown to decrease expression of additional important epithelial marker genes, including Keratin 19 (KRT19) and adherens junctions components EVA1 and PTPRM, leading to increased cell motility. This indicates that as PDAC progresses, lower GLI1 levels may actually prime tumor cells towards an EMT program, which would be associated with metastasis and advanced stages of the disease.
Mills et al. examined the role of GLI1 expression in the later stages of PDAC using a mouse model for advanced pancreatic cancer (Mills et al., 2014). In this study, the loss of GLI1 actually accelerated PDAC progression during the later stages of tumorigenesis. PDAC mice lacking GLI1 showed reduced survival when compared to GLI1 wild type littermates. While both cohorts of mice displayed the common features of advanced PDAC, loss of GLI1 was associated with decreased survival and increased tumor burden. Analysis of the mechanism revealed the pro-apoptotic FAS/FASL axis as a potential mediator for this phenomenon. Loss of GLI1 was associated with a significant decrease in expression of FAS/FASL, leading to lower apoptosis levels and increased tumor progression (Figure 1).
In agreement with these findings, two recent studies demonstrated that the deletion of the GLI1 inducer SHH, using a mouse model for PDAC, led to more aggressive tumors (Lee et al., 2014; Rhim et al., 2014). Interestingly, Rhim’s study reported the occurrence of poorly differentiated tumors, with increased vascularity, and significantly reduced stromal content. In contrast, the Lee paper only described a modest reduction in the stromal compartment. The current paradigm for PDAC is that the tumor stroma plays an important role in promotion of neoplastic growth and progression since PDAC is typically associated with a dense desmoplastic reaction. However, the Rhim study shows that tumors with reduced stroma may display a more aggressive behavior than those with an extensive stromal compartment. This concept is further supported by a recent report demonstrating that tumor stroma restrains pancreatic cancer progression and that pharmacological HH pathway activation in stromal cells can actually slow down in vivo tumorigenesis (Lee et al., 2014). The complexity of these findings reflects our incomplete understanding of the precise biological role of HH/GLI1 signaling in pancreatic cancer. In fact, the level of activation of HH signaling might induce different biological responses during the carcinogenesis process, as commonly observed during embryonic development. In fact, manipulation of the membrane mediators of HH signaling to reduce HH signaling leads to an increase of angiogenesis with low HH levels, but not with complete inhibition. Intriguingly, the Rhim and Lee studies generated a low HH signaling environment by eliminating SHH, but not IHH, another HH ligand expressed in pancreatic cancer. Similarly, studies altering the expression of Gli1 leave intact the other mediators of HH signaling, GLI2 and GLI3 (the latter mainly an inhibitor of HH target genes). It remains to be seen if manipulating GLI1 levels within the epithelial tumor compartment in later stages of disease is of any therapeutic value. Based on work from Fendrich et al. on HH signaling and acinar cell differentiation, it might even be provocatively proclaimed that increasing GLI1 levels could drive terminal differentiation and thus result in lower tumorigenicity (Fendrich et al., 2008).
Discussion
These studies demonstrating GLI1 may act as a tumor suppressor in the late stage of PDAC give insight into the disappointing results of clinical trials testing HH inhibitors in metastatic PDAC patients. While the Olive experiments reported acute administration of IPI-926 increased survival due to decreased stromal content and increased vascularity, the HH inhibitor performed poorly in pancreatic cancer clinical trials in patients. One explanation for this discrepancy may be the short duration of treatment (3 weeks) in the Olive’s experiments, which may have not accurately detected disease progression following HH inhibition. This indicates that as PDAC progresses, the initial positive effects of HH inhibition may be eliminated as GLI1 levels decrease. In Rhim’s study, the authors discovered that SHH and GLI1 deficient tumors were more aggressive, poorly differentiated, and exhibited increased vascularity (Rhim et al., 2014). This suggests HH/GLI1 pathway inhibition may have a proangiogenic effect. Due to the increase in vascularity of the SHH deficient mice tumors, the authors investigated the effect of angiogenesis inhibition by administering anti-VEGF to tumor-bearing SHH deficient mice. This therapy led to a significant improvement in the overall survival of mice with undifferentiated tumors. Based on this response, the subset of PDAC patients with undifferentiated tumors may benefit from anti-angiogenic therapy. In summary, due to the high complexity of PDAC initiation and progression, a personalized strategy for treatment should be considered. Under this strategy, PDAC should be analyzed before treatment to determine expression of GLI1 and upstream regulators in order to better define therapeutic options.
Author contributions
T.L.H and M.E.F.-Z. developed the concept of the review and T.L.H, M.L., M.P.M. and M.E.F.-Z. wrote the manuscript.
Competing interests
No competing interests were disclosed.
Grant information
This work was supported by National Institutes of Health Grant CA136526, Division of Oncology Research (Mayo Clinic), Mayo Clinic Pancreatic SPORE P50 Grant CA102701 and Mayo Clinic Center for Cell Signaling in Gastroenterology Grant P30 DK84567 (to M.E.F.-Z.).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Emily Porcher and Pam Becker for secretarial assistance.
Faculty Opinions recommendedReferences
- Algul H, Adler G, Schmid RM:
NF-kappaB/Rel transcriptional pathway: implications in pancreatic cancer.
Int J Gastrointest Cancer.
2002; 31(1–3): 71–78. PubMed Abstract
| Publisher Full Text
- Catenacci D, Bahary N, Sreenivasa RN, et al.:
Final analysis of a phase IB/randomized phase II study of gemcitabine (G) plus placebo (P) or vismodegib (V), a hedgehog (Hh) pathway inhibitor, in patients (pts) with metastatic pancreatic cancer (PC): A University of Chicago phase II consortium study.
J Clin Oncol.
2013; 31: (suppl; abstr 4012). Reference Source
- Dennler S, Andre J, Alexaki I, et al.:
Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo.
Cancer Res.
2007; 67(14): 6981–6986. PubMed Abstract
| Publisher Full Text
- Dennler S, Andre J, Verrecchia F, et al.:
Cloning of the human GLI2 Promoter: transcriptional activation by transforming growth factor-beta via SMAD3/beta-catenin cooperation.
J Biol Chem.
2009; 284(46): 31523–31531. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Eberl M, Klingler S, Mangelberger D, et al.:
Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells.
EMBO Mol Med.
2012; 4(3): 218–233. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Feldmann G, Dhara S, Fendrich V, et al.:
Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers.
Cancer Res.
2007; 67(5): 2187–2196. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Feldmann G, Habbe N, Dhara S, et al.:
Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer.
Gut.
2008; 57(10): 1420–1430. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Fendrich V, Esni F, Garay MV, et al.:
Hedgehog signaling is required for effective regeneration of exocrine pancreas.
Gastroenterology.
2008; 135(2): 621–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Fiaschi M, Rozell B, Bergström A, et al.:
Development of mammary tumors by conditional expression of GLI1.
Cancer Res.
2009; 69(11): 4810–4817. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Goel HL, Pursell B, Chang C, et al.:
GLI1 regulates a novel neuropilin-2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation.
EMBO Mol Med.
2013; 5(4): 488–508. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Hingorani SR, Wang L, Multani AS, et al.:
Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice.
Cancer Cell.
2005; 7(5): 469–83. PubMed Abstract
| Publisher Full Text
- Hui CC, Angers S:
Gli proteins in development and disease.
Annu Rev Cell Dev Biol.
2011; 27: 513–537. PubMed Abstract
| Publisher Full Text
- Javelaud D, Pierrat MJ, Mauviel A:
Crosstalk between TGF-β and hedgehog signaling in cancer.
FEBS Lett.
2012; 586(14): 2016–2025. PubMed Abstract
| Publisher Full Text
- Ji Z, Mei FC, Xie J, et al.:
Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells.
J Biol Chem.
2007; 282(19): 14048–14055. PubMed Abstract
| Publisher Full Text
- Jones S, Zhang X, Parsons DW, et al.:
Core signaling pathways in human pancreatic cancers revealed by global genomic analyses.
Science.
2008; 321(5897): 1801–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Joost S, Almada LL, Rohnalter V, et al.:
GLI1 inhibition promotes epithelial-to-mesenchymal transition in pancreatic cancer cells.
Cancer Res.
2012; 72(1): 88–99. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Kinzler KW, Bigner SH, Bigner DD, et al.:
Identification of an amplified, highly expressed gene in a human glioma.
Science.
1987; 236(4797): 70–73. PubMed Abstract
| Publisher Full Text
- Lauth M, Bergstrom A, Shimokawa T, et al.:
DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS.
Nat Struct Mol Biol.
2010; 17(6): 718–725. PubMed Abstract
| Publisher Full Text
- Lee JJ, Perera RM, Wang H, et al.:
Stromal response to Hedgehog signaling restrains pancreatic cancer progression.
Proc Natl Acad Sci U S A.
2014; 111(30): E3091–3100. PubMed Abstract
| Publisher Full Text
| Free Full Text
- LoRusso PM, Rudin CM, Reddy JC, et al.:
Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors.
Clin Cancer Res.
2011; 17(8): 2502–2511. PubMed Abstract
| Publisher Full Text
- Mahadevan D, Von Hoff DD:
Tumor-stroma interactions in pancreatic ductal adenocarcinoma.
Mol Cancer Ther.
2007; 6(4): 1186–1197. PubMed Abstract
| Publisher Full Text
- McMahon AP, Ingham PW, Tabin CJ, et al.:
Developmental roles and clinical significance of hedgehog signaling.
Curr Top Dev Biol.
2003; 53: 1–114. PubMed Abstract
| Publisher Full Text
- Mills L, Zhang DL, Marler R, et al.:
Inactivation of the transcription factor GLI1 accelerates pancreatic cancer progression.
J Biol Chem.
2014; 289(23): 16516–25. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Mills LD, Zhang Y, Marler RJ, et al.:
Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation.
J Biol Chem.
2013; 288(17): 11786–11794. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Nolan-Stevaux O, Lau J, Truitt ML, et al.:
GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation.
Genes Dev.
2009; 23(1): 24–36. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Nye MD, Almada LL, Fernandez-Barrena MG, et al.:
The transcription factor GLI1 interacts with SMAD proteins to modulate transforming growth factor β-induced gene expression in a p300/CREB-binding protein-associated factor (PCAF)-dependent manner.
J Biol Chem.
2014; 289(22): 15495–506. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Olive KP, Jacobetz MA, Davidson CJ, et al.:
Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer.
Science.
2009; 324(5933): 1457–1461. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Ougolkov AV, Fernandez-Zapico ME, Savoy DN, et al.:
Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells.
Cancer Res.
2005; 65(6): 2076–2081. PubMed Abstract
| Publisher Full Text
- Pan X, Arumugam T, Yamamoto T, et al.:
Nuclear factor-kappaB p65/relA silencing induces apoptosis and increases gemcitabine effectiveness in a subset of pancreatic cancer cells.
Clin Cancer Res.
2008; 14(24): 8143–8151. PubMed Abstract
| Publisher Full Text
- Rajurkar M, De Jesus-Monge WE, Driscoll DR, et al.:
The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis.
Proc Natl Acad Sci U S A.
2012; 109(17): E1038–1047. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Rhim AD, Oberstein PE, Thomas DH, et al.:
Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma.
Cancer Cell.
2014; 25(6): 735–747. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Seidel B, Braeg S, Adler G, et al.:
E- and N-cadherin differ with respect to their associated p120ctn isoforms and their ability to suppress invasive growth in pancreatic cancer cells.
Oncogene.
2004; 23(32): 5532–5542. PubMed Abstract
| Publisher Full Text
- Thayer SP, di Magliano MP, Heiser PW, et al.:
Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis.
Nature.
2003; 425(6960): 851–856. PubMed Abstract
| Publisher Full Text
| Free Full Text
- von Burstin J, Eser S, Paul MC, et al.:
E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex.
Gastroenterology.
2009; 137(1): 361–371, 371.e1–5. PubMed Abstract
| Publisher Full Text
- Wang W, Abbruzzese JL, Evans DB, et al.:
The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells.
Clin Cancer Res.
1999; 5(1): 119–127. PubMed Abstract
- Yauch RL, Gould SE, Scales SJ, et al.:
A paracrine requirement for hedgehog signalling in cancer.
Nature.
2008; 455(7211): 406–410. PubMed Abstract
| Publisher Full Text
Comments on this article Comments (0)