Keywords
α-synuclein, transcriptional regulation, evolutionary conserved genomic region, SNCA, Parkinson’s disease
α-synuclein, transcriptional regulation, evolutionary conserved genomic region, SNCA, Parkinson’s disease
We made corrections and edits according to the reviewers comments and addressed all questions and concerns in the body of the manuscript and Tables, and made changes in Figures 2 and 3.
See the authors' detailed response to the review by Ornit Chiba-Falek and Lidia Tagliafierro
An emerging hypothesis is gaining increasing interest and is based on the concept that subtle overexpression of α-synuclein (α-syn) over many decades can either predispose or even cause the neurodegenerative changes that characterize Parkinson’s disease (PD). Neurons subjected to higher, non-physiological levels of α-syn might be more likely to be damaged by oligomerization or aggregation of this protein, eventually leading to the formation of α-synuclein-based neuropathological features of the disease1.
It is now well established that both point mutations and large genomic multiplications of the α-syn (SNCA) gene can cause an autosomal-dominant form of PD2–10. Furthermore, several association studies investigating genetic variants in the SNCA gene have found an increased risk for PD11–19. The finding that both qualitative and quantitative alterations in the SNCA gene are associated with the development of a parkinsonian phenotype indicates that amino acid substitutions as well as overexpression of wild-type α-syn are capable of triggering a clinicopathological process that is very similar to sporadic PD. Nevertheless, the precise mechanisms leading to α-syn-related pathology in sporadic PD in the absence of any α-syn mutations remain elusive.
The best characterized polymorphism in the SNCA gene is the Rep-1 mixed dinucleotide repeat which has been shown to act as a modulator of SNCA transcription14–16. The DNA binding protein and transcriptional regulator PARP-1 showed specific binding to SNCA-Rep1. These data were confirmed by a transgenic mouse model and demonstrated regulatory translational activity20.
Functionally, SNCA expression levels in postmortem brains suggest that the Rep-1 allele and SNPs in the 3′ region of the SNCA gene have a significant effect on SNCA mRNA levels in the substantia nigra and the temporal cortex21.
The promoter region of the SNCA gene has been recently examined in more detail in cancer cell lines and also in rat cortical neurons. Regulatory regions in intron 1 and the 5′ region of exon 1 have been shown to exhibit transcriptional activation22–24 as well as the NACP-Rep-1 region upstream of the SNCA gene14–16,20,25. Several transcription factors have been identified such as PARP-116, GATA26, ZIPRO1, and ZNF21922 to have an effect on regulating the SNCA promoter region.
There is mounting evidence that SNCA expression levels could be crucial for maintenance and survival of neurons and its misregulation could play a key role in the development of PD. Thus, the importance of thoroughly investigating the SNCA gene to fully understand its cis- and trans-acting elements and factors and for the functional interpretation of the PD-disease associated risk alleles is becoming increasingly clear.
The goal of this study was to investigate transcriptional regulation of the SNCA region using a complementary approach, under the hypothesis that conserved non-coding regions of the SNCA gene are comprised of transcriptional enhancers or silencers and thus modulate gene expression. This would mean that single nucleotide polymorphisms (SNPs) in these regions could influence the transcriptional pattern of the SNCA gene27.
Using comparative genomics, we searched for highly conserved non-coding sequences between human and mouse and identified 34 evolutionary conserved non-coding genomic regions (ncECRs) within the SNCA gene that are conserved between human and mouse.
We utilized two complementary browsers (Vista browser (http://pipeline.lbl.gov/cgi-bin/gateway2) and ECR browser (http://ecrbrowser.dcode.org/) to generate a conservation profile by aligning the human SNCA gene with its mouse counterpart in a pair-wise fashion. We applied established selection parameters for our search with >100bp in length and >75% identity28,29. In addition to the 111.4kb SNCA gene region, we included a 44.5kb upstream and a 50kb downstream intergenic region to also capture surrounding regulatory elements.
We identified 34 ncECRs in the SNCA genomic region of 206kb on chromosome 4q21 (Chr.4: 90,961056-91,167082, UCSC Genome Browser on Human Mar. 2006 Assembly) by pair-wise comparison between human and mouse (Figure 1). Ten of these DNA sequences were located downstream of the SNCA gene, 17 were intronic between exon 4 and 5, which is 92kb in length, and five were upstream of the SNCA gene (Figure 1). None of the selected sequences overlapped with known expressed sequence tags (ESTs) or had an open reading frame of more than 20 amino acids in length, suggesting that these ncECRs are non-coding.

Panel shows human-mouse pair-wise comparison of Human genome May 2004 and Mouse Sept. 2005. Pink marked peaks represent ncECRs, turquoise marked peak represent the untranslated region (UTR) of SNCA, blue marked peaks represent exons. D1-D10 are conserved regions downstream of SNCA. In1-In17 are intragenic conserved regions, and U1-U4-2/3 are upstream of SNCA. The black arrow on top shows the transcription orientation.
To test, if the ncECRs exhibit enhancer or silencer activity, we cloned all identified regions in specific reporter vectors and measured their luciferase activity after transfection into neuroblastoma cells. For our studies, we used the pGL3 luciferase reporter vectors (Promega, Cat. No. E1751, E1741, E1771, E1761) and the human neuroblastoma cell line SK-N-SH. NcECRs identified through the comparative analysis (Supplementary Table 1) were cloned upstream of a SV-40 promoter in the pGL3 promoter construct, transfected in SK-N-SH cells and assayed with the Dual-Luciferase® Reporter Assay System (Promega, Cat. No. E1910).
Some of these regions were combined in one vector because of their close proximity to each other. Primers with specific restriction sites (KpnI, BglII or XhoI from New England Biolabs Inc.) were designed to amplify the conserved elements, and PCR products with specific restriction sites were directly cloned into the pGL3 promoter vector to ensure correct orientation of the genomic elements (Supplementary Table 1). All constructs were sequenced to ensure that no point mutations were introduced through the amplification and/or cloning process.
For transfection experiments, we used a 96-well format (Nunc, Cat. No. 167008). Cells were plated one day before transfection at a density of 3000–5000 cell/well to reach 90–95% confluency at the time of transfection, luciferase assays were performed 24hrs after transfection. SK-N-SH cells were maintained in Hyclone DMEM media (High Glucose, Fisher Scientific, Cat No. SH30081.02) with 10% Hyclone fetal bovine serum (Fisher Scientific, Cat No. SH30910.03) in 1× glutamine (Life Technologies, Cat. No. 25030-081) and 1× penicillin/streptomycin (Life Technologies, Cat. No. 15140-122). For SK-N-SH cells, we used 1:2 ratio of nucleic acid to transfection reagent (Lipofectamine® 2000 Transfection Reagent, Life Technologies, Cat No. 11668-019). For the luciferase assay, we used the Dual-Luciferase® Reporter (DLR™) Assay System (Promega, Cat. No. E1910) according to the manufacturer’s instructions in 96-well white plates, flat bottom (E&K Scientific, Cat. No. EK-25075). In this assay, activities of firefly and Renilla luciferases were measured sequentially in one sample. All assays were performed in quadruplicate and each experiment was repeated three times. Altogether, 12 data points were ascertained for each conserved region/construct.
Differences among means were analyzed using two-samples student’s t-test. For differences in transcriptional activation of the luc+ gene, ncECRs were tested in quadruplicates in three independent experiments. Differences were considered statistically significant at p<0.05.
To estimate the number of potential TFBSs and the number of interacting transcription factors (TFs) that could represent potential candidate proteins for our positive ncECRs, we used MatInspector in an in silico approach. We chose two elements for this bioinformatic analysis with MatInspector. The MatInspector software utilizes a large library of matrices for TFBSs to locate matching DNA sequences. The program assigns quality rating to matches and allows quality-based filtering and selection of matches. MatInspector can group similar or functionally related TFBSs into matrix families30.
In addition to the original human-mouse comparison, we added the sequences for dog and cow for comparisons. Only the TFBSs were considered that were present in all four species, in the same orientation, and similar distance to each other. We ran two analyses with 10 and 15 nucleotides distance, respectively. We accepted only models in which at least four TFs can bind in a concerted way. Each TFBS can potentially bind several TFs.
We also computationally tested all possible TFs for interactions with the SNCA promoter region, which were retrieved from the proprietary ElDorado database (Genomatix, Munich, Germany). In this database, promoters are defined and ranked by transcription start sites, corresponding known mRNA or EST sequences and by orthologous conservation.
Overall, 12 of 34 conserved non-coding elements exhibited either an increase or reduction of the expression of the luciferase reporter gene (Figure 2 and Dataset 1). Three elements upstream of the SNCA gene (U3, U4-1, and U4-3) displayed a significant approximately 1.5 fold (p<0.009) increase in expression (Figure 2A). Of the intronic regions, three showed a 1.5 fold increase (I2, I6, I8) and two others showed a 2 and 2.5 fold increase in expression (p<0.002), I5 and I12, respectively (Figure 2B). Two elements downstream of the SNCA gene showed approximately 2 fold (D1 and D2) and 2.5 fold (D3) increase (p<0.0009) (Figure 2C). One element D6 downstream of SNCA had a reduced expression of the reporter gene of 0.35 fold (p<0.0009) of normal activity (Figure 2C, green) that was also confirmed after cloning the D6 element in a pGL3 control vector (Figure 2C, insert). The pGL3 control vector contains the SV-40 promoter and a SV-40 enhancer element. The D6 element reduced the expression of the pGL3 control construct by ~50%, confirming that this element represents a repressor. Between 4 and 12 replicates were performed per ncECR.
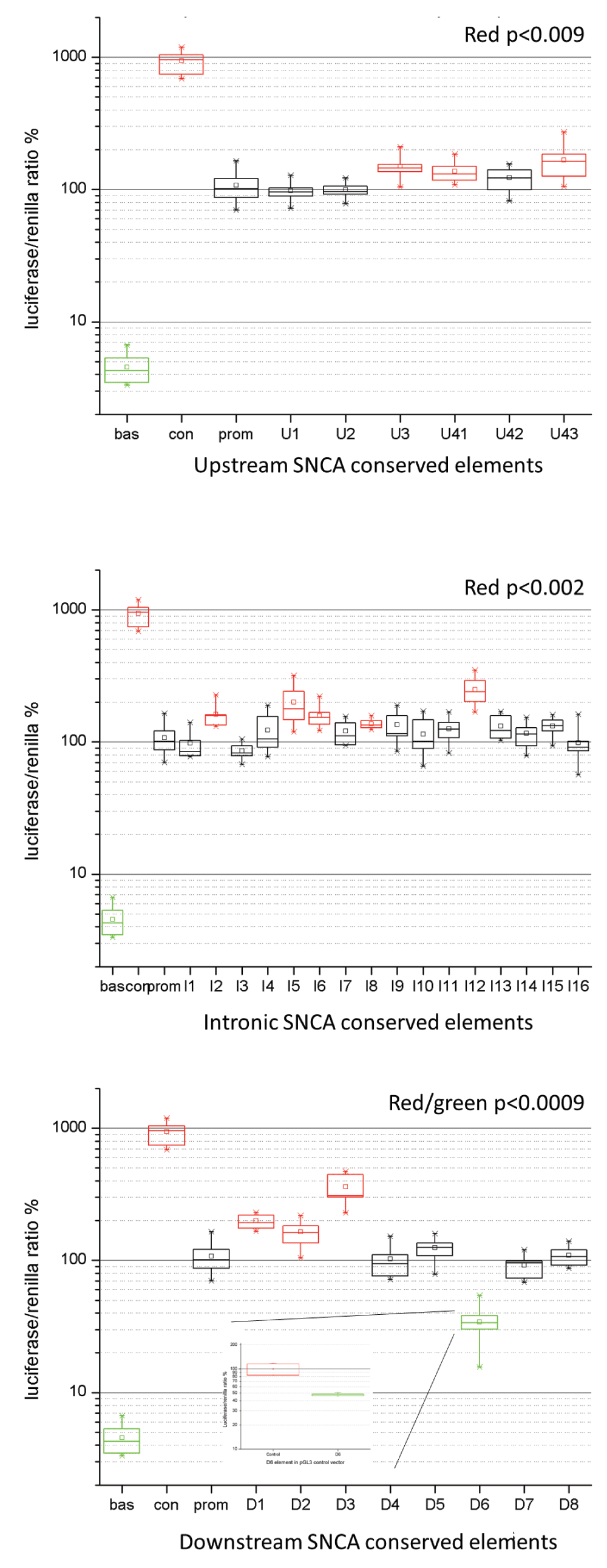
Panels A–C show the luciferase assay results of ncECRs upstream (A), intragenic (B), and downstream (C) of the SNCA gene. The X-axis shows the ncECRs, the Y-axis shows the ratio of luciferase and renilla expression as percentage. Bas=pGL3 basic, Con=pGL3 control, prom=pGL3 promoter construct. All red or green box plot elements represent ncECRs that modulate expression significantly. The box plots show the median (horizontal line within box), the 25 and 75% tiles (horizontal borders of box), and the whiskers show the minimal and maximal values. Panel C, insert: Luciferase assay results of D6 element cloned into the pGL3 control vector construct.
These data provide experimental evidence that a significant proportion of the ncECRs show a regulatory function in the luciferase reporter assay.
We performed MatInspector (Genomatix) analysis30 on two elements (I12:chr4:90940532-90940786 and D6: chr4:90855871-90856339, Human Genome assembly NCBI36/hg18) with the highest fold change in the luciferase assay. In addition to the original human-mouse comparison to identify the ncECRs, we added the sequences from dog and cow. Only TFBSs that were present in all four species, in the same orientation, and similar distance to each other were considered. We ran two analyses with 10 and 15 nucleotides distance, respectively. We accepted only models in which at least four TFs can bind in a concerted way. Each TFBS can potentially bind several TFs. Interestingly, using this more restricted model, five factors showed an interaction with the SNCA promoter as well as with the ncECRs (Figure 3A). These factors were the Paired-like homeodomain transcription factor 3 (PITX3), the Homolog of Drosophila orthodenticle 2 (OTX2), the Nuclear receptor subfamily 3, group c, member 1 (NR3C1) or glucocorticoid receptor (GCCR), the Androgen receptor (AR), and the general transcription initiation factor TATA box-binding protein (TBP).

A. MatInspector network view of SNCA promoter interaction with TFs that also potentially bind to two ncECRs (I12 and D6) within the SNCA gene. M=gene product is part of metabolic pathway, IN=input gene, TF=transcription factor, ST=gene product is part of signal transduction pathway, green line=matches target promoter B. UCSC Genome browser custom track of PD associated SNPs (based on PD Gene metaanalysis), Rep1 allele and functional ECR regions on chromosome 4 (Human Genome Assembly Feb. 2009, GRCh37/hg19).
It is intriguing to note that by searching for TFs that bind to both the promoter and the functional ncECR, several DNA-binding proteins were found that are linked to dopaminergic regulation and susceptibility for nigrostriatal impairment. Two of these TFs (PITX3 and OTX2) implicated in determination of a dopaminergic phenotype in the substantia nigra emerged from this preliminary search31,32. PITX3 has shown to be regulated in a negative feedback circuit through the microRNA mi-133b to fine-tune maintenance of dopaminergic neurons33. In an association study, a SNP in the PITX3 promoter was reported to be associated with PD and might dysregulate expression of PITX334 suggesting that transcription factors play a critical role not only in the development and differentiation of dopaminergic neurons, but also for cell maintenance and survival of dopaminergic neurons.
GCCR and AR belong to a class of nuclear receptors called activated class I steroid receptors. GCCR is a cytosolic ligand-activated transcription factor that regulates the expression of glucocorticoid-responsive genes. GCCR shows strong anti-inflammatory and immunosuppressive effects. Interestingly, impaired GCCR expression in a mouse model shows a dramatic increase in the vulnerability of the nigrostriatal dopaminergic neurons to a toxic insult of MPTP35.
Taken together, this preliminary in silico screen resulted in very intriguing new candidates that might directly regulate SNCA expression and could play a role in the pathological processes that underlie PD.
A major focus in PD research has been on post-translational modification of α-syn. The alterations seen in PD that were linked to disease pathogenesis were nitrated α-syn and α-syn phosphorylated at serine 129 identified in Lewy bodies and Lewy neurites36,37, however, the gene transcription as a control point and its regulation in particular cell types or upon cellular signals has only been touched fairly recently in PD-relevant genes.
Our results show that potential regulatory regions are not restricted to the promoter of the SNCA gene as discussed in the introduction, but are likely to be located also in other intronic and intergenic regions (Figure 3B). Comparing our results to similar screens, where conserved regions range from 8–45 elements38,41, we found a similar number of functional elements in our screen that show a high evolutionary conservation.
Not only the promoter region of a gene drives the transcription/expression of a gene. Also other cis-acting genomic regions within a certain gene, up to several hundred kb away, can serve as enhancers, silencers, or modifiers to ensure the accurate temporal and spatial expression of a gene by recruiting transcription activating or silencing factors that bind to them38. There is ample precedence for this approach to analyze genomic regions of genes implicated in human disease. Mutations in those conserved elements were found to cause human genetic syndromes, for example SALL1/Townes-Brocks syndrome39 or SHH/preaxialpolydactyly40. Other groups have investigated the non-coding regulatory elements within disease genes such as RET (Ret proto-oncogene) and MECP2 (Methyl-CpG binding protein 2) and found multiple regulatory enhancer and silencer elements38,41.
Computationally determining transcription factor binding sites is a challenging process and multiple prediction algorithms have been developed over the last decade (Cartharius 2005, Wu 2009, Mathelier 2013). Therefore our preliminary data should solely open the discussion and drive novel hypotheses for potential transcription factors that regulate transcription of the SNCA locus. Specific TFs seem to be directly involved in neurodegeneration and models of PD. TFs have been shown to be critical regulators for the development, maintenance and survival of dopaminergic neuronal populations42,43. E.g. forkhead transcription factor (Foxa2) is responsible for early development of endoderm and midline structures. Foxa2 is specifically expressed in postmitotic dopaminergic neurons. Genetically engineered mice that are null for Foxa2 are not viable, whereas heterozygotes for Foxa2 develop major motor abnormalities starting at 18 months with an asymmetric posture, rigidity, and bradykinesia44.
This screen of evolutionary conserved genomic elements in the SNCA locus showed a number of functionally elements that in an in vitro assay modulated the expression of a reporter gene. Furthermore, we identified very intriguing new candidate transcription factors that could directly regulate SNCA expression and could, if binding is altered by genetic variants, play a role in the pathological processes that underlie PD. This is the first step to systematically analyze the SNCA locus to understand its transcriptional regulation in more detail. Further studies are needed in neuronal tissues (e.g. dopaminergic neurons derived from patient-specific induced pluripotent stem cells) to confirm these findings and expand the analysis to identify SNCA-regulating transcription factors. By defining the transcription factors that regulate expression and potentially overexpression of α-synuclein that can lead to neurodegeneration, we will be able to identify targets for novel therapeutic approaches for α-synucleinopathies including Parkinson’s disease.
F1000Research: Dataset 1. Combined normalized raw datasets of Luciferase assays on SNCA conserved elements, http://dx.doi.org/10.5256/f1000research.3281.d3745245
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)