Introduction
Oxidative stress is usually considered a mechanism of brain aging1. However, contradictory data2 and lack of firm evidence3 makes it difficult to firmly establish its actual significance in this process (see López-Otín et al.4 for a recent review). One important aspect that requires further clarification in this regard is the relationship between oxidative stress and insulin peptides, a well conserved family of hormones firmly linked to aging. Extensive work in vertebrates and invertebrates indicates that the insulin-like growth factor I (IGF-I)/insulin signalling (IIS) pathway has a negative impact on aging. It has been argued that this detrimental action is mediated by reducing cell defences to oxidative stress5–7 which, in turn is harmful for neuronal survival1. However, IGF-I has been shown to be largely neuroprotective8, even in conditions such as ischemic injury or brain trauma where oxidative stress is most likely a major pathogenic mechanism9. Thus, it is unclear whether or not IGF-I protects the brain against oxidative stress as the current evidence is contradictory.
A possible explanation for these apparently contradictory observations may be that modulation of the cellular response to oxidative stress by IGF-I is cell-dependent10. Until now, only neurons have been studied in this regard. However, astrocytes, a major cellular element of the brain, are essential contributors to neuronal homeostasis and are coupled to neurons in the response to oxidative stress in order to help protect them11. It is thus possible that IGF-I participates in the response of astrocytes to oxidative stress as part of the overall brain response encompassing all types of brain cells, not only neurons. Contrary to what we previously observed in neurons12, we report here that IGF-I protects astrocytes against oxidative stress and, very significantly, also co-operates with astrocytes to protect neurons.
Methods
Animals
We used postnatal rats and mice for in vitro cultures (P0-3 days for astrocytes and P7 for neurons) and 3 month old mice for in vivo experiments. P2 Wistar rats (8 g ± 0.04 body weight, n=240, Harlan, Spain), P3 (2 g ± 0.03, n=36, Harlan) and 3 months old (27.6 g ± 0.812; n= 24) C57BL6 mice and P7 GFP transgenic mouse pups (4.25 g ± 0.22, n=126; in-house colony) were used. Pups used were of both sexes and no attempt to sex them was done. Adult mice were male. Rat tissue was used in all in vitro experiments except when using GFP cell derived from transgenic mice. In vivo experiments were done in mice for future comparisons with transgenic mice. All efforts were made to minimize suffering and reduce the number of animals. Animals were kept under light/dark, 12 h/12 h) conditions following EU guidelines (directive 86/609/EEC) and handled according to institutionally-approved procedures (CSIC bio-ethics subcommittee project code SAF2010-1703). Animals were fed ad libitum with laboratory rodent chow (Teklad Global 2018S) and kept in standard laboratory cage conditions with 4 animals/cage.
Reagents
Antibodies used in this study are detailed in Table 1. The different drug inhibitors used in the study are given in Table 2. Hydrogen peroxide (H2O2) and the calcium chelator BAPTA-AM were purchased from Sigma (Steinheim, Germany). IGF-I and SCF were purchased from Prospec-Tany Technogene, (Israel).
Table 1. Antibodies used in the study.
| ANTIBODY | PRODUCT
Nº | MANUFACTURER | WORKING
CC | SPECIES | ISOTYPE | ANTIGEN
(EPITOPE) | AFFINITY
PURIFIED | REFERENCE |
|---|
| Akt1/2 (H-136) | sc-8312 | Santa Cruz
Biotechnology
(California, USA) | 1:1000 | rabbit | polyclonal | aminoacids
345–480 of
human Akt1/2 | unknown | 42–45 |
β-actin (Clone
AC-74) | A5316 | Sigma (Steinheim,
Germany) | 1:50000 | mouse | monoclonal | N-terminal end
of β-isoform
of actin | ascytes
fluid | 46–49 |
Cu/Zn
superoxide
dismutase
(SOD) | SOD-101 | Assay Designs
(Michigan, USA) | 1:1000 | rabbit | polyclonal | Native rat
Cu/Zn SOD | yes | 50,51 |
MnSOD
superoxide
dismutase
(SOD) | SOD-111 | Assay Designs
(Michigan, USA) | 1:2500 | rabbit | polyclonal | Native rat Mn
SOD | yes | 52–55 |
p44/p42 MAPK
(ERK1/2) | 9102 | Cell Signalling
(Danvers, USA) | 1:2000 | rabbit | polyclonal | sequence in
the C-terminal
of rat p44MAPK | yes | 56–59 |
phospho-Akt
(Ser473) | 9271 | Cell Signalling
(Danvers, USA) | 1:1000 | rabbit | polyclonal | residues
surrounding
Ser473 of
mouse Akt | yes | 46,60–63 |
phospho-
ERK1/2
(Thr202/Tyr204) | 9101 | Cell Signalling
(Danvers, USA) | 1:2000 | rabbit | polyclonal | residues
surrounding
Thr202/Tyr204
of human p44
MAPK | yes | 63,63,63–66 |
| SCF | sc-9132 | Santa Cruz
Biotechnology
(California, USA) | 1:1000 | rabbit | polyclonal | aminoacids
26–214 of
human SCF | unknown | 67–69 |
| TXNIP1 | K0205-3 | MBL (Nagoya,
Japan) | 1:2000 | mouse | monoclonal | human
recombinat
TXNIP | unknown | no refs |
Table 2. Drug inhibitors used in the study.
| Target | Inhibitor | Dose | Supplier |
|---|
| CALCINEURIN | CYCLOSPORIN A | 500 nM | Sigma-Aldrich |
| ERK MAPK | U0126 | 20 µM | Tocris Bioscience |
| Extracellular Ca2+ | CdCl2/EGTA | 100 µM/10 mM | Sigma-Aldrich |
| IGF-IR | PPP | 120 nM | Calbiochem |
| Intracellular Ca2+
| BAPTA/AM | 5–10 µM | Calbiochem |
| JNK | InsolutionTM JNK INHIBITOR II | 10–20 µM | Calbiochem |
| mTOR | InsolutionTM RAPAMYCIN | 100 nM | Calbiochem |
| NF-KB | QNZ | 10–20 nM | Enzo Life Sciences |
| p38 MAPK | SB203580 hydrochloride | 20 µM | Calbiochem |
| PDK1 | OSU-03012 | 10 µM | Echelon |
| PI3K | LY294002 | 25 µM | Calbiochem |
| PKA | KT5720 | 60 nM | Tocris Bioscience |
| PKC/PKA | Ro 31-8220 | 20–900 nM | Calbiochem |
| PKCα, PKCβI, PKCε | Rho 32-0432 | 0.2 µM | Calbiochem |
| PKC isotypes (α,β,γ,δ,ζ,µ) | Go6983 | 6 nM–20 µM | Tocris Bioscience |
| PP1 | TAUTOMYCIN | 2 nM | Calbiochem |
| PP2A | OKADAIC ACID (495609 Insolution) | 2.5 nM | Calbiochem |
| PROTEASOME | MG-132 | 5 nM–3 µM | Calbiochem |
| PROTEIN SYNTHESIS | CYCLOHEXIMIDE | 1 µg/ml | Calbiochem |
Plasmids
pECE-FOXO3 and pECE-FOXO3-TM (triple mutant T32A/S253A/S315A, herein called MFOXO3) were kindly provided by ME Greenberg (Harvard Medical School, Boston, USA). p6xDBE-luc (reporter luciferase plasmid with six copies of the DAF16 family protein-binding element) and pRL-TK (TK-Renilla luciferase) were a kind gift of BM Burgering (University Medical Centre, Utrecht, The Netherlands). Dominant negative IGF-IR expression plasmid was kindly donated by D. Le Roith (Mt Sinai, New York, USA). Plasmids expressing shRNA for TXNIP1 were purchased from Origene (USA). Txnip1 plasmid was purchased from Thermo Scientific Open Biosystems (Waltham, USA).
Cell culture and transfections
Cerebellar granule cultures were produced from either P7 rat or GFP transgenic mouse cerebella as previously described13. In brief, cells were plated onto 6 or 12-well dishes coated with poly-l-lysine (1 μg/ml) at a respective final density of 1.5×106/well or 0.45×106/well. Cells were incubated at 37°C/5% CO2 in Neurobasal (Gibco, USA) medium supplemented with 10% B27 (Gibco), glutamine (5 mM) and KCl (25 mM). All experiments were carried out in 2–7 day old cultures, with neurons showing neurite extensions. Different times in vitro were used to analyze time-dependent parameters such as cell survival. Rat granule neurons were transfected 24 h after plating. The DNA: transfection agent ratio (Neurofect, Genlantis, San Diego, USA) was 1:7. The percentage of neurons transfected was 5–10%, as assessed with a GFP vector. Neurons were left untreated for at least 48 hours. On the day of the experiment, medium was replaced with Neurobasal + 25 mM KCl. Two hours later, IGF-I (10-7 M) and/or hydrogen peroxide (H2O2) at doses of 50–150 µM were added. Inhibitory drugs were given 45 min before treatments. We used H2O2 as an oxidant stimulus because it is an endogenously produced reactive oxygen species (ROS) that serves as a precursor to hydroxyl radicals and possesses signalling capacities14. Astrocyte cultures were prepared from P3 rat or GFP mouse forebrain, as previously described15 after animals were sacrificed by decapitation. Cells were grown on Dulbecco’s modified Eagle's medium F12 (DMEM-F12) supplemented with 10% fetal calf serum. After 12 days astrocytes were seeded at 2.5×105 or 1.25×105 cells/well in 6-well and 12-well culture plates, respectively. On the day of the experiment cells were treated with IGF-I (10-7M), H2O2 (50–200 µM) and/or inhibitors, as above. For transfection, astrocytes were seeded at 2.5×105 or 1.25×105 cells/well in 6-well and 12-well culture plates respectively, and after 16 h constructs were mixed with Fugene HD (Roche, Switzerland) in a 1:3 ratio, and added following the manufacturer’s instructions. Alternatively, astrocytes were electroporated (2×106 astrocytes with 2 µg DNA or shRNA) before seeding using an astrocyte Nucleofector Kit (Lonza, Switzerland). After electroporation, cells were plated to obtain a final cell density on the day of the experiment similar to that obtained with the transfection method. All experiments were performed after 48 h. The transfection efficiency was 20–30% and 60–80% for electroporation, as assessed with a GFP vector. At least three independent experiments were done in duplicate wells.
Co-cultures
For co-cultures, 1.25×105 wild type mouse astrocytes/well were seeded on 12-well plates and grown with DMEM-F12 plus 10% FBS. After 48–72 hours, GFP neurons were isolated and plated onto astrocytes. We used forebrain astrocytes and cerebellar neurons because in our experience the forebrain and cerebellum yielded very high numbers of astrocytes and neurons, respectively (thus minimizing animal use). Furthermore, in this study we were interested in exploring general, rather than region-specific neuroprotective characteristics of astrocytes. Nevertheless, we also carried out co-cultures with neurons and astrocytes from the same region (forebrain) and the results obtained were identical than when using cells from differing regions (see Figure 2 in results). Culture medium was changed to DMEM-F12 plus B27, 4 mM glutamine and 25 mM KCl (the latter only in the case of neurons). Two days later, co-cultures were treated with 100 nM IGF-I ± 50–100 µM H2O2. Pictures were taken every 24 hours up to 5–7 days as above. For protein silencing or overexpression, 2×106 astrocytes were electroporated in a Nucleofector®II (Amaxa Biosystems Lonza, Switzerland) and seeded at 1.25×105/well. Co-cultured neurons were seeded as described above. Viability of neurons was assessed by counting the number of cells expressing GFP using Incucyte software (2010A) with a set cell size threshold to avoid including GFP+ cell debris and dying cells. This threshold ranged from 8–36 µm2 to 70–200 µm2 depending on the experiment. Viability is expressed as percentage of GFP+ cells at the beginning of the experiment (time 0). At least three independent experiments were done.
Cell assays
Cell viability was determined by four different methods. The first assessed astrocyte death by quantification of the amount of lactate dehydrogenase (LDH) released from damaged astrocytes into the culture medium. LDH levels were measured after 16 h of treatment with different H2O2 concentrations using a commercial kit (Roche Diagnostics, Germany). When using transfected astrocytes, a GFP-pCMV vector and the different constructs under evaluation were used in a 1:5 ratio. In this case, GFP+ astrocytes were scored prior to treatment to determine baseline survival (time 0) and at different times as indicated in the results. Alternative viability assays for astrocytes included measuring cell metabolism with fluorescein diacetate (0.1 µg/ml FDA) or number of propidium iodide (PI) cells12 as specified in the results section. For the latter, astrocytes or neurons were stained with 2 µg/ml PI as a marker of dead cells plus DAPI staining as a marker of total cell number. PI+ and DAPI+ cells were counted under a Leica CTR 6000 fluorescence microscope. Percentage of viable cells indicates the number of PI+ cells related to total cell number. The experiments were done in triplicate and a total of three independent experiments were done. For neuronal-specific viability assays cerebellar neurons from GFP mice were seeded on 12-well plates (4.5×105 cells) coated with poly-L-Lysine and grown with Neurobasal medium plus B27, 4 mM glutamine and 25 mM KCl. After 4–5 days, cultures were treated with 100 nM IGF-I in the presence or absence of 50–100 µM H2O2. Pictures of GFP+ cells (green fluorescence) were taken every 24 hours up to 3 days in an IncucyteTM 2010A Rev2 system (Essen BioScience, USA). Viability of neurons in co-culture experiments was measured as described above.
Immunoassays
Western blotting was performed as described13. Cells were washed once with ice-cold PBS and lysed with 1% NP-40, 150 mM NaCl, 20 mM Tris, pH 7.4, 10% glycerol, 1 mM CaCl2, 1 mM MgCl2, 400 μM sodium vanadate, 0.2 mM PMSF, 1 µg/ml leupeptin, 1 µg/ml aprotinin and 0.1% phosphatase inhibitor cocktails I and II (Sigma-Aldrich). To normalize for protein load, membranes were reblotted (Re-Blot, Chemicon, USA) and incubated with an appropriate control antibody (see Results). Levels of the protein under study were expressed relative to protein load. Different exposures of each blot were collected to ensure linearity and to match control levels for quantification.
Densitometric analysis was performed using Analysis Image Program (Bio-Rad, USA). A representative blot is shown from a total of at least three independent experiments. IGF-I levels in culture medium were measured using Quantikine ELISA for mouse/rat IGF-1 (R&D Systems, USA). In brief, cells were treated as described above and 1 ml of culture medium was collected after 24 hours, spun to eliminate cell debris, and stored at -80ºC. Samples were lyophilized overnight and resuspended in 150 µl of calibrator buffer. After vortexing, samples were centrifuged 10 min/14,000 rpm (Hettich, Germany) and assayed according to manufacturer’s instructions. A total of three and four independent experiments were done for neurons and astrocytes, respectively. SCF levels in culture medium were measured by western blot after collecting the supernatants and processing them as described above for IGF-I. After lyophilisation, samples were resuspended in western blot lysis buffer and protein levels were measured by Bradford (Biorad, Germany) following the manufacturer’s instructions to normalize for protein load in SDS-PAGE gels.
Luciferase assays
Luciferase assays were done as previously reported12. In brief, cells were transfected with a reporter construct bearing six canonical FOXO binding sites (6×DBE- luciferase) and co-transfected with different constructs, as indicated in each experiment. Transfections were performed in triplicate dishes. Luciferase counts were normalized using TK-Renilla luciferase. At given times, neurons were lysed in passive lysis buffer (PLB) and luciferase activity was analysed using a luminometer and dual luciferase assay kit according to the manufacturer (Promega, USA). Background luminescence was subtracted. Luciferase activity was expressed as fold of increase over control levels. At least three independent experiments were done.
Flow cytometry
After 18h of exposure to H2O2, cell death was assessed. Cells were detached using 0.25% Trypsin-1.3 mM EDTA (Invitrogen) during 5–10 minutes, centrifuged (200×g, 5 min/4ºC), and resuspended in cold PBS. Propidium iodide (PI 5 µg/ml; Sigma) in PBS was added prior to flow cytometry analysis using a FACSAria cytometer (BD Biosciences). Fluorescence intensity, forward scatter (FSC), and side scatter (SSC) were collected in logarithmic scale. The emission filter used was 600–620 nm band pass (FL3). A fluorescence blank was measured and subtracted from the fluorescence of the sample. Dead cells were identified as red fluorescence positive events with low FSC (small PI permeable cells). Debris was always excluded from the analysis. At least three independent experiments were conducted.
ROS measurement
Mitochondrial O2- production levels were measured by using the fluorescent probe MitoSOXTM Red (Life Technologies, USA). Briefly, astrocytes were pre-treated overnight with IGF-I and then 200 µM H2O2 were added during 1 hour. Cells were incubated with 1.5 µM MitoSOXTM Red in DMEM-F12 for 10 min/37ºC and washed 3 times with PBS. Astrocytes were then trypsinized and fluorescence was measured by flow cytometry (510 nm excitation/580 nm emission) using the cytometer, as described15. A total of six independent experiments were done. Alternatively, ROS generation was assessed in astrocytes cultured on coverslips with the fluorogenic marker carboxy-H2DCFDA (Molecular Probes, USA) during 30 min/37ºC, protected from the light. When using this ROS marker it is not possible to distinguish endogenous ROS from exogenously applied H2O2. Nevertheless, we compared this method to the oxidation of luminol (which detects superoxide anions) that distinguishes H2O2 from other ROS and we obtained identical results with either method (data not visualized). The reason we used carboxy-H2DCFDA is because we could obtain both qualitative (cell images) and quantitative (fluorimetry assay) measurements within the same assay. After incubation with carboxy-H2DCFDA, cells were gently washed 3 times with warm DMEN, and mounted, or, alternatively, lysed for fluorimetry. Pictures were taken at 40× magnification using a Leica fluorescence microscope (Germany). A representative picture is shown. Fluorescence intensity in lysed cells was measured using a FluoroStar fluorimeter.
Growth factor gene array
An RT2 ProfilerTM PCR Array (SABiosciences, USA) was used to screen a battery of growth factors following the manufacturer’s recommendations. After treatment, astrocytes were lysed and RNA extracted using Trizol (Life Technologies, USA). The resulting cDNA synthesis reaction was diluted in water, mixed with the qPCR master mix, and loaded in a 96 well PCR Array plate. PCR was performed following manufacturer’s instructions.
Brain focal ischemia
Three-month old male mice (4–6 per group) were anesthetized with 3% isoflurane (in 70% N2O, 30% O2) for induction and with 2% isoflurane for maintenance. Rectal temperature was maintained at 36.5 C with a heating pad. The frontal branch of the medial cerebral artery (MCA) was exposed and occluded permanently by suture ligation as previously reported, with modifications16. Briefly, an incision perpendicular to the line connecting the lateral canthus of the left eye and the external auditory canal was made to expose and retract the temporalis muscle. A burr hole was drilled, and frontal and parietal branches of the MCA were exposed by cutting and retracting the dura. The frontal branch of the MCA was elevated and ligated with a suture nylon monofilament 8/0. Following ligation, a sharp decrease of blood flow was evidenced with a laser Doppler flowmetry (Järfalla, Sweden). Following surgery, mice were returned to their cages, kept at room temperature and allowed free access to food and water. All physiological parameters measured: rectal temperature, mean arterial pressure and blood glucose levels were not different between groups. Sixteen hours after medial cerebral artery occlusion (MCAO), animals were killed by neck dislocation by an experienced researcher to assess infarct outcome. The brain was removed and the infarcted area isolated and processed for RNA and protein isolation.
Quantitative PCR
Total RNA isolation from cell lysates or brain tissue was carried out with Trizol. One µg of RNA was reverse transcribed using High Capacity cDNA Reverse Transcription Kit (Life Technologies) according to the manufacturer’s instructions. For the quantification of specific genes, total RNA was isolated and transcribed as above and 62.5 ng of cDNA was amplified using TaqMan probes for Txnip1, IGF-I or SCF and 18S as endogenous control (Life Technologies). Each sample was run in triplicate in 20 μl of reaction volume using TaqMan Universal PCR Master Mix according to the manufacturer’s instructions (Life Technologies). All reactions were performed in a 7500 Real Time PCR system (Life Technologies). Quantitative real time PCR analysis was carried out as previously described17. Results were expressed as relative expression ratios on the basis of group means for target transcripts versus reference 18S transcript. At least three independent experiments were done.
Statistical analysis
Data are expressed as mean ± SEM. Differences among groups were analyzed by one- or two-way ANOVA followed by a Newman-Keul’s or Student’s t-test using Graph Pad Prism 5 software. A p<0.05 was considered significant.
Results
Astrocyte neuroprotection against oxidative stress requires IGF-I signalling onto astrocytes
Whereas neurons cultured without astrocytes are very sensitive to acute oxidative insult elicited by H2O2 (Figure 1A), when cultured with astrocytes, neurons become very resilient (Figure 1A). To determine whether IGF-I participates in the neuroprotective effects of astrocytes against oxidative stress we first confirmed that it is endogenously produced by these cells. As shown in Figure 1B, not only astrocytes but also neurons (albeit at much lower levels) secrete IGF-I into the culture medium. In response to H2O2 astrocytes secrete lower, but still substantial, amounts of IGF-I, and so IGF-I may still participate in neuroprotection by astrocytes. To directly test this possibility we blocked IGF-I signalling in astrocytes with a dominant negative (DN) IGF-IR18 (Figure 1C) and determined their ability to protect neurons against oxidative challenge. As shown in Figure 1D, a significantly greater percentage of neurons co-cultured with mock-transfected astrocytes survived after H2O2 challenge than when cultured with astrocytes transfected with DN IGF-IR.
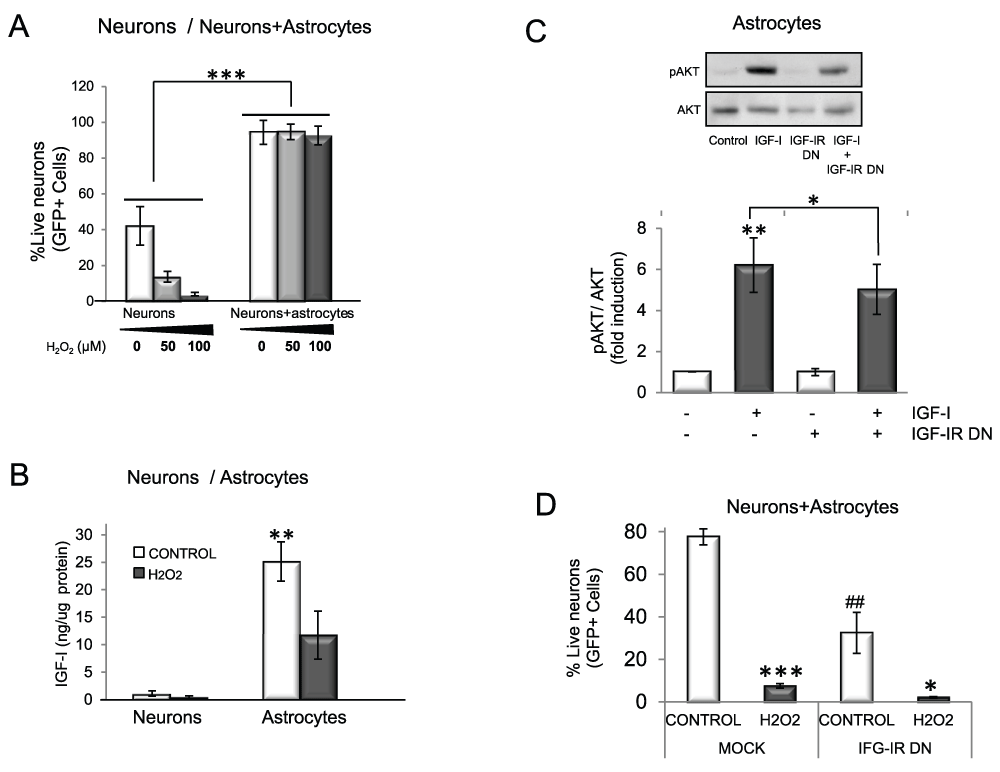
Figure 1. IGF-I signalling participates in astrocyte neuroprotection against oxidative injury.
A) Neurons are protected from oxidative stress in the presence of astrocytes whereas when cultured alone they rapidly die. Viability of GFP neurons was measured as the number of green (GFP+) cells two days after H2O2 treatment in the presence or absence of wild type astrocytes (F=41.85; ***p<0.001 vs. neurons alone. B) Both astrocytes and neurons secrete IGF-I, although astrocytes produce much higher levels (*p<0.05 vs neurons). H2O2 lowers IGF-I secretion. C) In the presence of a dominant negative IGF-IR (IGF-IR DN) signalling by IGF-I was markedly reduced. Astrocytes were transfected with IGF-IR DN or mock transfected, and the ratio pAkt/Akt (histograms) was measured as an index of IGF-I signalling. Representative blots and quantitative histograms are shown (2 way ANOVA, IGF-I and IGF-IR DN interaction: p<0.05, F=6.99; IGF-I p<0.01, F=13.46; IGF-IR DN p<0.05, F=7.06; Post-hoc: **p<0.01 vs control (mock-transfected) and *p<0.05 vs. IGF-I+IGF-IR DN). D) Blockade of IGF-IR function with IGF-IR DN compromises neuroprotection by astrocytes. GFP neurons were seeded on top of wild type astrocytes transfected with an IGF-IR DN construct or mock-transfected (control) and exposed to 100 µM H2O2. Viability of GFP neurons was measured after 5 days (2 way ANOVA, H2O2 and IGF-IR interaction: p<0.05, F=10.77; H2O2 p<0.01, F=68.92; IGF-IR DN p<0.05 F=17.86; post-hoc: ***p<0.001, *p<0.05 vs. Control; ##p<0.01 vs mock). Experiments were done at least 3 times in this and following figures. Bars are SEM in all figures.
We next used pharmacological blockade of the IGF-I receptor using picropodophyllin (PPP), an antagonist of IGF-IR (Figure 2A). As in this case both the neuronal and astrocyte receptors are blocked, we first determined whether neurons are affected by PPP blockade of the IGF-I receptor. In the presence of H2O2, neurons cultured alone die regardless of the presence or absence of proper IGF-I signalling since PPP did not increase neuronal death (Figure 2B). This agrees with our previous findings that IGF-I does not protect cultured neurons against oxidative stress12. Confirming the results seen with astrocytes transfected with dominant negative IGF-I receptor, a reduction in neuroprotection by astrocytes was seen when co-cultured neurons were exposed to PPP. In the presence of H2O2, significantly fewer co-cultured neurons survived with PPP (p<0.01 H2O2+PPP vs. H2O2 alone; Figure 2C). To rule out region-specific actions of astrocytes on neuroprotection we then co-cultured neurons and astrocytes from the same brain region (forebrain) and treated them with PPP. As shown in Figure 2D, forebrain neurons were similarly sensitive to blockade of IGF-IR when co-cultured with forebrain astrocytes. The observation that even supra-physiological doses of IGF-I (100 nM) added to the co-cultures only produced a modest additional effect on neuronal survival after oxidative insult confirmed the idea that endogenous IGF-I is required by astrocytes for neuroprotection (Figure 2E). Hence, endogenous production of IGF-I is necessary and sufficient to protect neurons.
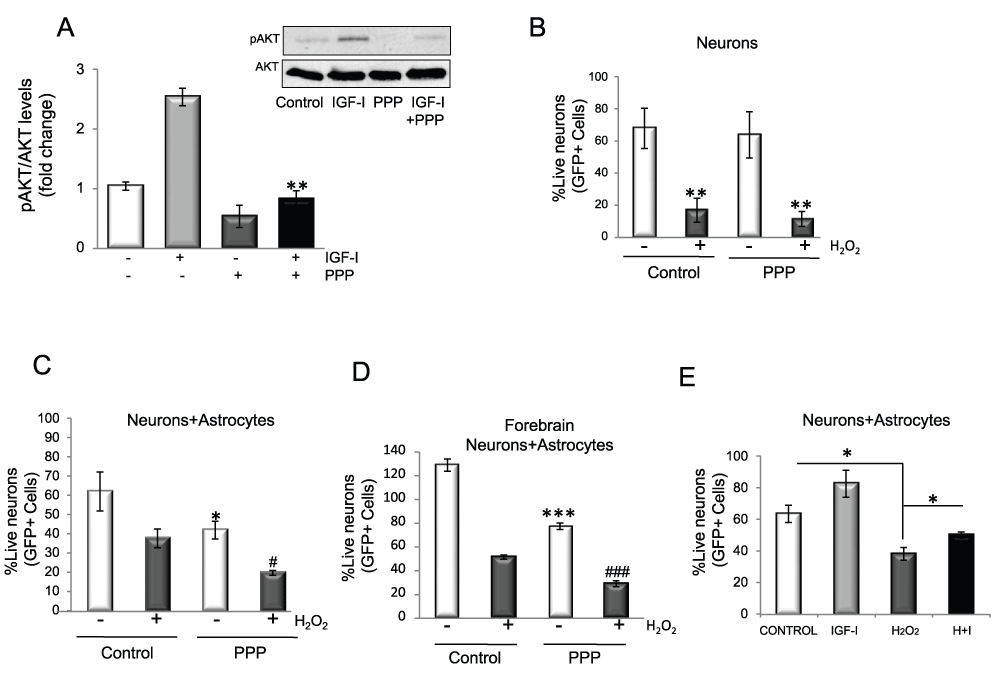
Figure 2. Endogenously produced IGF-I protects neurons against oxidative injury.
A) The IGF-IR inhibitor PPP blocks IGF-I signalling in astrocytes. Astrocytes were treated with 120 nM PPP 1h before adding IGF-I while pAkt levels were measured 10 minutes after adding IGF-I. Ratios are shown in histograms (2 way ANOVA, IGF-I and PPP interaction: p<0.01, F=33.07; IGF-I p<0.01, F=27.38; PPP p<0.001, F=112.3; post-hoc: **p<0.01 vs. IGF-I alone). Representative blot is shown. B) Blockade of IGF-IR signalling with PPP in neurons cultured alone does not affect H2O2 toxicity after 3–4 days of exposure (2 way ANOVA, H2O2 and PPP interaction: F=0.069; H2O2 p<0.01, F=12.43; PPP F=3.66; post-hoc: **p<0.01 vs. respective controls). Note that PPP alone does not affect neuronal survival. C) Viability of cerebellar neurons co-cultured with forebrain astrocytes decreased significantly when treated with PPP for six days. PPP treatment in the presence of H2O2 decreased neuronal viability even further (2 way ANOVA, H2O2 and PPP interaction: F=0.097; H2O2 p<0.05, F=9.65; PPP p<0.01, F=31.33; post-hoc: *p<0.05 vs untreated control and #p<0.05 vs H2O2). D) Viability of forebrain neurons co-cultured with forebrain astrocytes decreased significantly when treated with PPP for five days. PPP treatment in the presence of H2O2 decreased neuronal viability even further (2 way ANOVA, H2O2 and PPP interaction: p<0.001, F=23.74; H2O2 p<0.001, F=321.6; PPP p<0.001, F=151.3, post-hoc: ***p<0.01 vs. untreated control and ### p<0.01 vs. H2O2). E) When co-cultured with wild type astrocytes, neuronal survival after five days of exposure to 100 µM H2O2 was moderately increased in the presence of 100 nM IGF-I (2 way ANOVA, H2O2 and IGF-I interaction: F=0.542; IGF-I p<0.05, F=7.28; H2O2 p<0.001, F=25.9; post-hoc: *p<0.05 vs. control or H2O2). I+H: IGF-I + H2O2.
IGF-I protects astrocytes against oxidative stress
IGF-I-dependent neuroprotection by astrocytes appears to also involve a direct action of IGF-I on astrocytes. Because it is known that astrocytes are more resistant to oxidative damage than neurons, we explored whether IGF-I was involved in this greater resilience. Contrary to neurons (Figure 3A), IGF-I protected astrocytes against H2O2-induced death (Figure 3B). The protective effect of IGF-I involved blockade of the activation of FOXO 3, a transcription factor involved in brain responses to oxidative stress19, by H2O2 (Figure 3C). Inhibition of FOXO 3 by IGF-I was mediated by Akt; i.e.: an Akt-insensitive mutant of FOXO (M-FOXO3) abrogated IGF-I effects while wild type FOXO3 did not interfere with its protective actions (Figure 3D). Indeed, in astrocytes IGF-I activates Akt in the presence of H2O2 (Figure 3E), whereas in neurons H2O2 blocks this canonical pathway12. Underlying the protective actions of IGF-I on astrocytes was its ability to block excess ROS after exposure to H2O2 as determined by flow cytometry using MitoSOX (Figure 4A) or fluorometry with carboxy-H2DCFDA (Figure 4B).
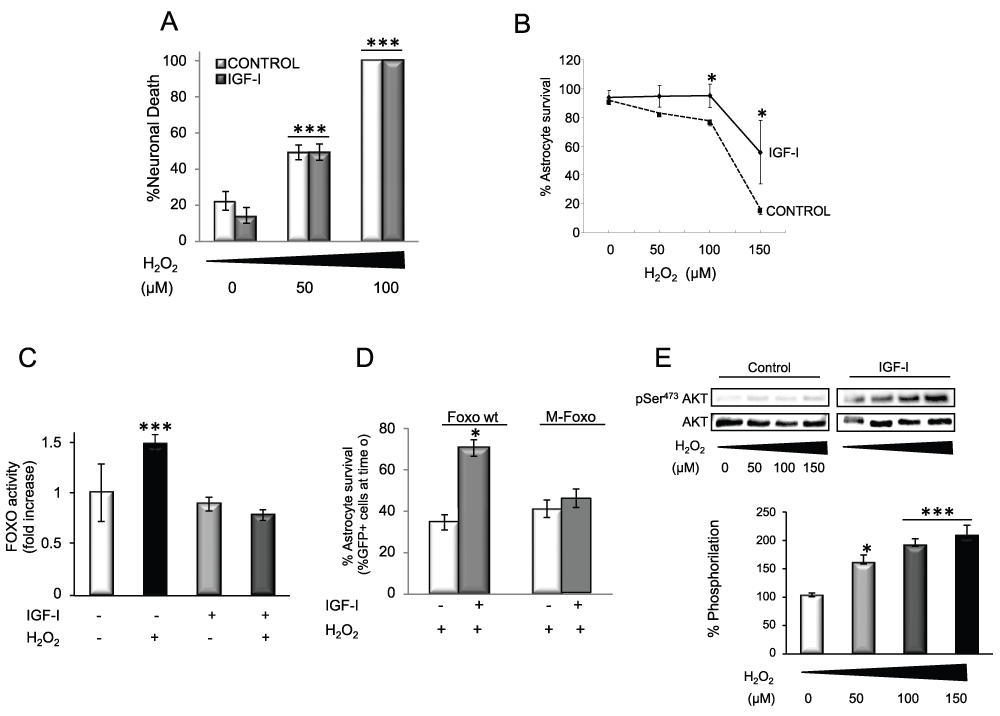
Figure 3. IGF-I protects astrocytes against oxidative stress.
A) Whereas IGF-I increases neuronal survival under control conditions, it does not protect neurons from H2O2 induced death. This confirms previous observations12. Neuronal mortality was measured by counting PI+ cells 6h after treatment. H2O2 induces neuronal death in a dose-dependent manner irrespective of the presence of IGF-I (2 way ANOVA, H2O2 and IGF-I interaction: p<0.001, F=10.3; IGF-I p<0.05, F=9.98; H2O2 p<0.001, F=128.7; post-hoc: ***p<0.001 vs. no H2O2¸, # p<0.05 vs control). B) IGF-I treatment protects astrocytes from H2O2 induced death. Astrocyte demise was measured by counting PI+ cells 24 h after H2O2 (100 µM). H2O2 exerts a dose-dependent effect that is reduced by IGF-I (2 way ANOVA, H2O2 and IGF-I interaction: p<0.01, F=5.36; IGF-I p<0.001, F=30.29; H2O2 p<0.001, F=60.42; post-hoc: *p<0.05 vs control). C) IGF-I blocks FOXO activity induced by H2O2 (100 µM). FOXO activity was measured with a luciferase reporter in astrocytes treated with IGF-I, H2O2 or both for 24 h (2 way ANOVA, H2O2 and IGF-I interaction: p<0.001, F=25.98; IGF-I p<0.001, F=49.58; H2O2 p<0.01, F=10.47; post-hoc: ***p<0.001 vs no treatment). D) Protection by IGF-I against cell death induced by H2O2 requires blockade of FOXO activity. Astrocyte viability was measured by counting GFP+ astrocytes after co-transfection of GFP and a FOXO wild type (wt) or an Akt-insensitive mutant of FOXO (M-FOXO; 2 way ANOVA, M-FOXO and IGF-I interaction: p<0.01, F=59.99; IGF-I p<0.05, F=13.31; M-FOXO p<0.01, F=21.84; post-hoc: *p<0.05 vs no IGF-I). E) IGF-I increases phosphorylation of Akt (pAkt) in the presence of H2O2 in a dose-dependent fashion. Representative blots are shown. Lower histograms indicate quantification of pAkt/Akt ratio in the presence of IGF-I as shown in the right blot. pAkt levels were measured after 15 min. (*p<0.05 and ***p<0.001 vs. no H2O2).
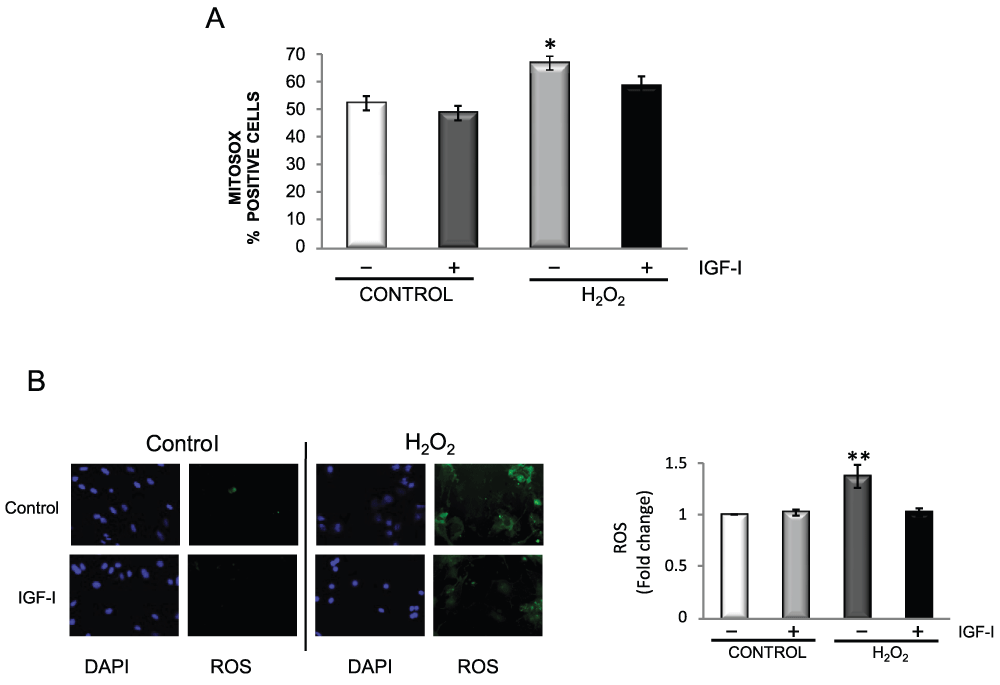
Figure 4. IGF-I reduces oxidative stress in astrocytes.
A) H2O2 increases the number of astrocytes expressing mitochondrial O2-. This increase is prevented when cells are pre-treated with IGF-I. Mitochondrial O2- levels were detected with MitoSOX by flow cytometry. Astrocytes were treated overnight with IGF-I and for 1 hour more with 200 µM H2O2 (2 way ANOVA, H2O2 and IGF-I interaction: F=1.27; IGF-I p<0.05, F=8.18; H2O2 p<0.01, F=16.18; post-hoc: **p<0.01 H2O2 vs control, *p<0.05 H2O2 vs IGF-I + H2O2). B) IGF-I lowers ROS levels after treatment of astrocytes with H2O2 (100 µM). Left: representative photomicrographs of astrocytes stained with carboxy-H2DCFDA to detect ROS and DAPI to stain cell nuclei. The increase in fluorescent cells elicited by H2O2 was markedly diminished by IGF-I. Right histograms: fluorimetric quantification of ROS levels with carboxy-H2DCFDA confirmed the rescuing action of IGF-I on astrocytes exposed to H2O2. (2 way ANOVA, H2O2 and IGF-I interaction: p<0.05, F=7.38; IGF-I p<0.05, F=5.89; H2O2 p<0.05, F=8.49; post-hoc: **p<0.01 H2O2 vs control, IGF-I, or IGF-I + H2O2).
We then determined possible mediators of the anti-oxidative actions of IGF-I on astrocytes. We examined whether modulation of SODs could be involved because these anti-oxidant enzymes constitute an important detoxifying mechanism in cases of excess ROS. We found that cytosolic Cu/ZnSOD was increased by IGF-I, H2O2, or both (Figure 5A), while mitochondrial MnSOD was increased only by H2O2 (Figure 5B). Thus, increases in SOD levels form part of the astrocyte response to H2O2, and IGF-I does not appear to interfere with these enzymes. Because FOXO participates in cellular responses to ROS, we looked for signals downstream of FOXO inactivation by IGF-I such as thioredoxin inhibitor 1 (TXNIP1), a pro-apoptotic protein dependant on FOXO activity and related to anti-oxidant responses20. We first confirmed that in astrocytes TXNIP1 is also controlled by FOXO; i.e.: in astrocytes expressing dominant negative Foxo, TXNIP1 levels were 89% reduced as compared to mock-transfected astrocytes. Accordingly, IGF-I, which inhibits FOXO, also reduced TXNIP1 levels (Figure 6A). Strikingly, H2O2, which stimulates FOXO activity in astrocytes (Figure 3D), also inhibited TXNIP1 (Figure 6A), suggesting alternative routes of TXNIP1 regulation in the presence of H2O2. When IGF-I and H2O2 were simultaneously added to astrocytes, TXNIP1 levels were markedly decreased (p<0.05 vs. IGF-I or H2O2 alone, Figure 6A). To determine the impact of downregulation of TXNIP1 on astrocyte survival we inhibited its expression with shRNA (blot in Figure 6B left panel) and found that astrocytes became resistant to H2O2 when TXNIP1 levels were low (Figure 6B). Overexpression of TXNIP1 did not alter the response of astrocytes to H2O2 whereas co-culture of neurons with astrocytes depleted of TXNIP1 did not result in enhanced neuronal survival (Figure 6B right panel), indicating that this route is involved in the response of astrocytes to oxidative stress but not in neuroprotection. Interestingly, in neurons, TXNIP1 was downregulated only in the presence of H2O2, but not after IGF-I treatment (Figure 6C). Thus, IGF-I down-regulates TXNIP1 only in astrocytes, not in neurons.
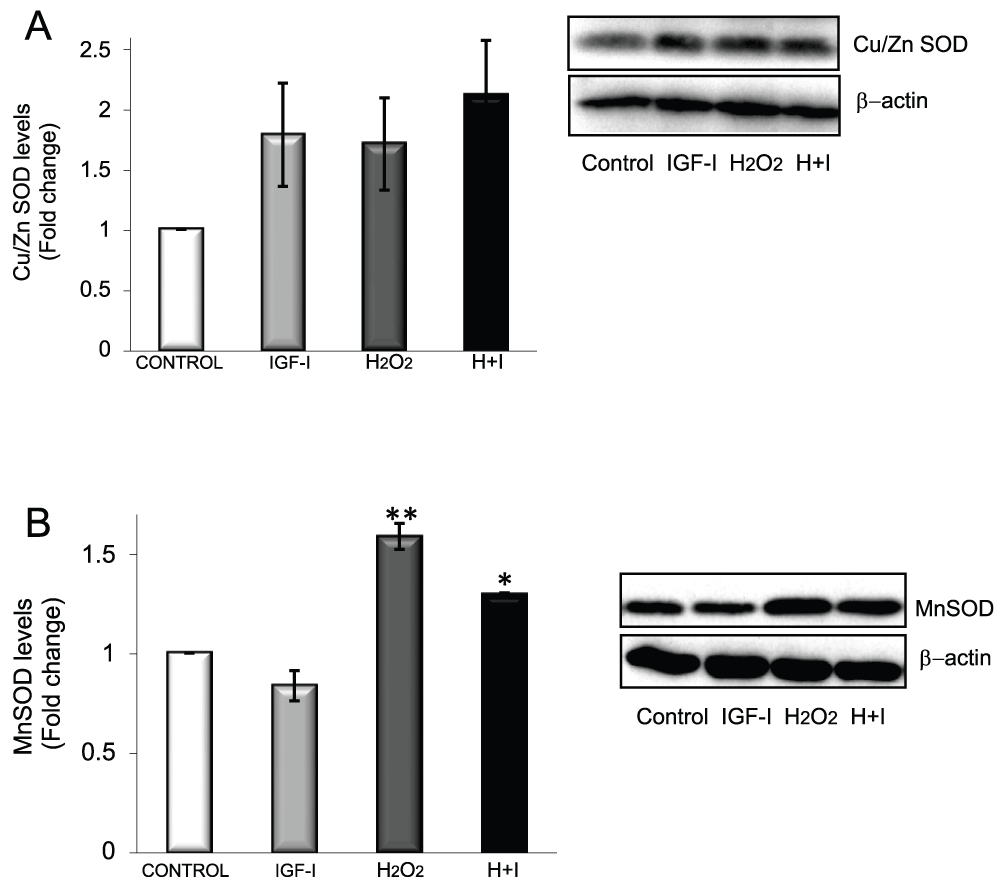
Figure 5. SOD responses to oxidative stress in astrocytes.
A) Cu/ZnSOD levels in astrocytes are modulated by IGF-I and H2O2. B) MnSOD levels are enhanced by H2O2 but not by IGF-I (*p<0.05 and **p<0.01 vs control).
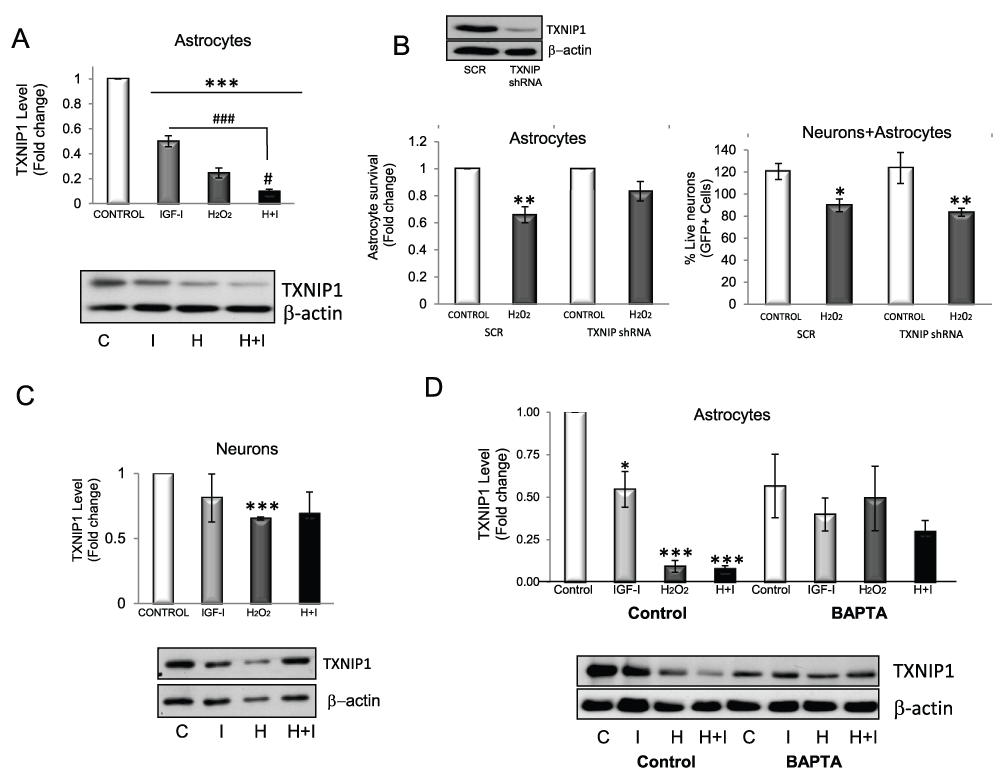
Figure 6. Both H2O2 and IGF-I reduce TXNIP1 in astrocytes.
A) Levels of the pro-oxidant protein TXNIP1 are reduced by IGF-I and H2O2. Inhibition is greater when both are added together (F=156.6; ***p<0.001 vs. control and ###p<0.001 (vs. IGF-I) and #p<0.05 (vs. H2O2). Levels of actin in each sample were measured to normalize TXNIP1 levels. B) Western blot: transfection of astrocytes with shRNA TXNIP1 results in reduced TXNIP1 levels as compared to astrocytes transfected with scrambled shRNA (SCR). Left panel: TXNIP1 shRNA silencing makes astrocytes less sensitive to H2O2 toxicity. Astrocyte viability was measured by FDA in the presence of 200µM H2O2 (2 way ANOVA, TXNIP1 and H2O2 interaction: F=2.94; TXNIP1 shRNA, F=2.94; H2O2, p<0.001, F=35.5; post-hoc: **p<0.01 vs control). Right panel: However, neuronal viability is not increased by reduced TXNIP1 in astrocytes as neurons die in the same proportion after H2O2 challenge. Viability of neurons was determined after co-culture for three days with astrocytes transfected with TXNIP1 shRNA (2 way ANOVA, TXNIP1 and H2O2 interaction: F=0.93; TXNIP1 shRNA, F=0.0097; H2O2; p<0.05, F=10.95). C) In neurons, only H2O2 decreases TXNIP1 levels, whereas IGF-I does not (***p<0.001 vs control). D) Reduction of TXNIP1 by IGF-I and H2O2 in astrocytes depends on Ca2+ as in the presence of the calcium chelator BAPTA-AM, the decrease is abrogated. (F=7.226; *p<0.05 and ***p<0.001 vs. control). C=control, I=IGF-I, H=H2O2, H+I=H2O2 + IGF-I.
We then analyzed possible pathways involved in the inhibitory effect of H2O2 and IGF-I on TXNIP1. Using kinase inhibitors we ruled out the idea that the main kinases downstream of the IGF-I receptor or H2O2 were involved. In fact, inhibition of most of these kinases resulted in altered basal levels of TXNIP1 (not visualized), suggesting that basal levels of this protein are tightly regulated in astrocytes. Other inhibitory drugs of different pathways where IGF-I participates (PKC, PKA, CnA, PDK-1, NFκB, among others) gave similar negative results. However, inhibition of Ca++ flux with 5 µM BAPTA abrogated TXNIP1 decreases in response to either H2O2 or IGF-I while only slightly, but not significantly affecting basal levels (Figure 6D).
IGF-I cooperates with SCF produced by astrocytes to protect neurons against oxidative stress
We next analyzed possible neuroprotective effects of IGF-I through astrocytes. Using a commercial gene array for growth factors we screened growth factor production by IGF-I-treated astrocytes in response to H2O2. Among the several growth factors that increased, stem cell factor (SCF) showed the highest elevation (Table 3). We confirmed by qPCR that SCF mRNA was increased after H2O2 whereas IGF-I decreased it (Figure 7A upper panel). Accordingly, levels of soluble SCF (sSCF) in culture medium from astrocytes treated with H2O2 were also increased (Figure 7A, lower panel). As SCF has been shown to be neuroprotective21, we determined whether it protects neurons against H2O2 and found that while SCF alone did not exert any protection, co-treatment with IGF-I resulted in significantly greater neuronal survival (p<0.05; Figure 7B). We then examined pathways underlying this cooperative action of IGF-I and SCF. Under basal conditions, the activity of extracellular signal-regulated kinase (Erk; measured as pErk/Erk ratio), a canonical kinase in IGF-I signalling, was increased by IGF-I as expected, and to a lesser extent also by SCF (Figure 7C). Basal Erk activity was also increased by H2O2. However, Erk was no longer activated by IGF-I or SCF in the presence of H2O2. Only when both were added together to H2O2-challenged cultures Erk activity was increased (Figure 7C). No interactions were found with Akt, the other canonical kinase pathway activated by IGF-I.
Table 3. Growth factors array.
| Upregulated genes | Downregulated genes |
|---|
Gene
symbol | Fold regulation | Gene
symbol | Fold regulation |
|---|
| Bmp4 | 34.1544 | Amh | -43.2611 |
| Bmp8a | 19.7667 | Bdnf | -5.4642 |
| IGF-I | 185.7219 | Bmp1 | -51.304 |
| IL7 | 54.0417 | Bmp2 | -8.3513 |
| Inhbb | 595.9304 | Bmp3 | -311.6969 |
| SCF | 3072.0799 | Bmp6 | -30.211 |
| VEGFb | 22.0239 | Bmp7 | -40.3361 |
| | Clcf1 | -7.5162 |
| | Csf1 | -6.4666 |
| | Csf3 | -102.9643 |
| | Cxcl1 | -35.4079 |
| | Cxcl12 | -49.3166 |
| | Egf | -24.916 |
| | Ereg | -10.8078 |
| | Fgf1 | -11.353 |
| | Fgf10 | -15.6273 |
| | Fgf14 | -12.7639 |
| | Fgf18 | -21.0245 |
| | Fgf2 | -9.6934 |
| | Fgf22 | -13.5011 |
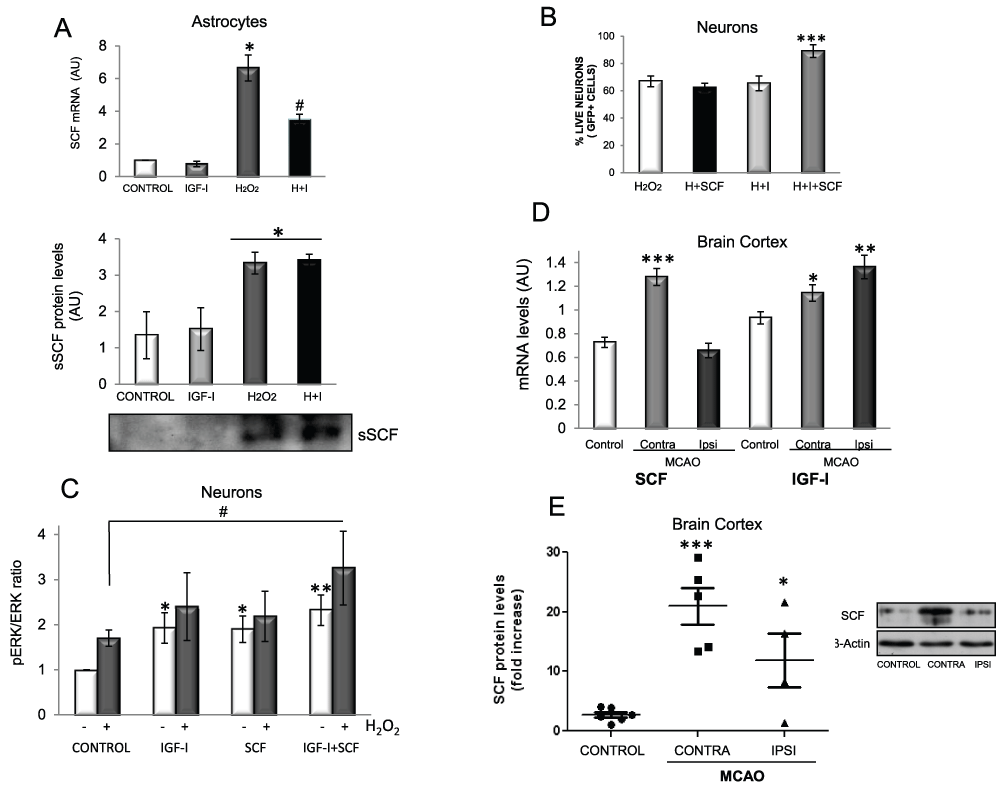
Figure 7. IGF-I cooperates with SCF to promote neuronal survival.
A1) Upper panel: H2O2 stimulates SCF mRNA levels in astrocytes after 16 h of exposure whereas IGF-I partially counteracts this increase (F=38.67; *p<0.05 vs. control and IGF-I, #p<0.05 vs. H2O2). A2) Lower panel: H2O2 stimulates SCF secretion. SCF levels in supernatants from astrocyte cultures treated or not with H2O2 and/or IGF-I for 24 h. A representative western blot is shown (*p<0.05 vs control). B) SCF and IGF-I cooperate to protect neurons from oxidative stress. Neurons were pre-treated with SCF, IGF-I or both 48 h before adding H2O2 (50 µM) and viability was assessed after overnight treatment (F=12.09, ***p<0.0001 vs H2O2), H: H2O2; I: IGF-I. C) When H2O2 is present, Erk phosphorylation is significantly increased only when both SCF and IGF-I are added to the cultures but not with either alone. Neurons were treated with 100 nM IGF-I, 20 ng/ml SCF and 50 µM H2O2 for 5 minutes and pErk levels were measured by western blot and normalized for total Erk. (*p<0.05 and **p<0.01 vs. control without H2O2 and #p<0.05 vs. H2O2). D) SCF and IGF-I mRNA levels increased 16 hours after middle cerebral artery occlusion (MCAO) in the contralateral side (CONTRA) in the case of SCF (F=31.53; ***p<0.001 vs. intact control mice) and in both sides in the case of IGF-I (F=7.853; *p<0.05 and **p<0.01 vs. control). E) SCF protein levels increase after MCAO in both sides of the cortex (F=12.38; *p<0.05 and ***p<0.001 vs. control). A representative blot is shown. Six, five and four animals were used per group, respectively. Levels of actin in each sample were measured to normalize for total protein levels.
To determine the in vivo relevance of these observations we submitted mice to brain ischemia as this brain insult is associated to oxidative stress22, and both IGF-I9 and SCF23 have been shown to be neuroprotective after ischemia. We found that IGF-I mRNA is increased after middle cerebral artery occlusion (MCAO) both in the ipsilateral and contralateral cortex, while only the contralateral side showed increased SCF mRNA levels compared to intact mice (Figure 7D). However, levels of SCF protein were elevated after MCAO in both the damaged and contralateral sides compared to normal mice (Figure 7E). This suggests that after brain ischemia the contralateral cortex produces higher amounts of SCF that eventually reach the ischemic side. Under this condition IGF-I may interact with SCF to promote neuronal survival in the ipsilateral cortex.
Discussion
The present results indicate that IGF-I exerts a protective action on astrocytes contributing to the resilience of these glial cells against oxidative stress. IGF-I also cooperates with astrocytes to protect neurons. These observations highlight the importance of cell-specific and cell-cooperative aspects of IGF-I protection against oxidative challenge. Thus, a better understanding of the trophic role of IGF-I in the brain requires taking into account its effects on astrocytes (and other brain cells) and the functional links of these cells with neurons. While these observations do not help settle the role of oxidative stress in brain aging they put forward an important aspect of possible mechanisms involved in aging; regulatory signals such as IGF-I may not modulate the response of the different cells and even tissues to oxidative stress in the same way.
The protection provided to astrocytes by IGF-I against oxidative stress may contribute to the greater resilience of these cells to oxidative challenge. In addition, astrocytes are coupled to neurons in the response to oxidative stress and provide them with ample detoxification support11. Among different anti-oxidant defences provided by astrocytes to neurons, we now find that IGF-I, which cannot protect isolated neurons against excess ROS12 cooperates with SCF secreted by astrocytes to support neurons (Figure 8). While in response to oxidative stress the production of IGF-I by cultured astrocytes and neurons is decreased, after brain ischemia IGF-I levels are actually higher due to increased synthesis and accumulation in microglia, vessels and astrocytes24. Therefore, in vivo, astrocytes and neurons will receive IGF-I input from various local sources, suggesting that the response of increased IGF-I after brain ischemia reflects an endogenous neuroprotective mechanism against oxidative injury. This conclusion apparently contradicts other evidence that IIS activity is pro-oxidant. Thus genetic ablation of ISS components in the nematode Caenorhabditis elegans25 or in higher organisms such as the fruit fly26 or mice27, increases organism resistance to oxidative stress. For example, mice with reduced IGF-I activity (hemizygous for the IGF-I receptor) have lower levels of ROS in the brain28,29. However, these mice developed greater cell damage after oxidative injury29. Conceivably, the effects of modulating IGF-I signalling prior to ROS insult (as when using genetic models) may not be the same compared to after insult. For example IGF-I protects nerve cells and/or the brain against diverse types of ROS-related insults30–34. In this regard, we recently reported that in a cellular model of Friedreich's ataxia (which elicits oxidative damage) neurons responded to IGF-I only when they became frataxin deficient, but not under normal conditions15. Collectively these observations emphasize the importance not only of cell type but also of context dependency of IGF-I neuroprotection in relation to oxidative stress.
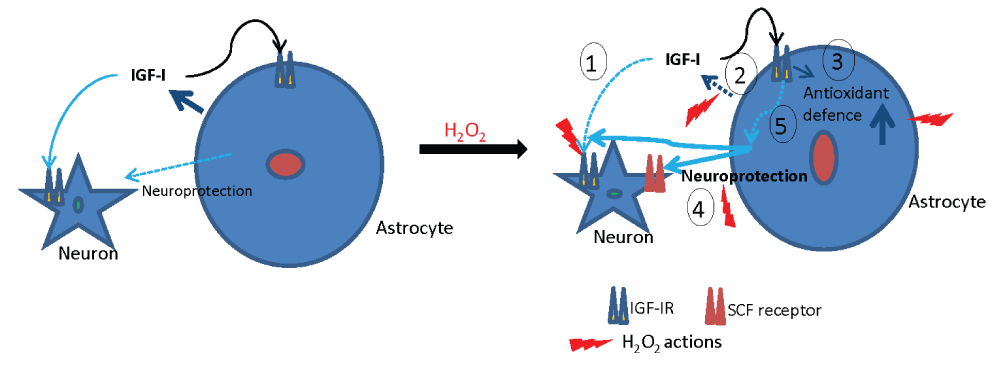
Figure 8. Schematic representation of IGF-I neuroprotection through astrocytes.
Left: under basal conditions IGF-I exerts potent neuroprotective actions directly onto neurons, as extensively documented previously8 (also shown in Figure 3A), and probably also through astrocytes. In the presence of H2O2 (right side) the actions of IGF-I on neurons and astrocytes can be summarized in 5 points: 1) IGF-I loses its ability to directly protect neurons, 2) IGF-I secretion by astrocytes is diminished, 3) IGF-I reinforces astrocyte defences against oxidative stress by down-regulating pro-oxidant mechanisms such as TXNIP1. 4) IGF-I cooperates with SCF secreted by astrocytes to promote neuronal survival. 5) However, the precise mechanism(s) downstream of astrocyte IGF-I receptors underlying enhanced astrocyte neuroprotection remains to be determined. Cytotoxic effects are depicted in red while cytoprotective actions are indicated in blue trace.
A role for oxidative stress in many neurodegenerative diseases is gaining increasing acceptance35. Aberrant production of ROS in the central nervous system is linked to neurodegenerative diseases such as Alzheimer’s dementia, Parkinson’s disease or stroke, all of them associated to aging36. However, as already commented, the role of oxidative stress in brain aging is still unclear. An attempt to explain these apparently opposing observations is that moderate ROS levels may activate survival pathways37. The present findings agree with this proposal. Thus, doses of H2O2 up to 100 µM do not elicit astrocyte death probably because IGF-I helps maintain their anti-oxidant capacity. In this regard our results show that astrocytes in response to IGF-I and/or H2O2 activate antioxidant signalling including upregulation of Cu/ZnSOD and MnSOD coupled to downregulation of pro-oxidant proteins such as Txnip1. Txnip1 inhibits thioredoxin (Trx), a protein that reduces protein disulfides as well as H2O2. The Txnip-Trx axis plays an important role in different brain diseases in which oxidative stress is implicated38.
There is ample evidence that different trophic factors, including SCF39, contribute to reduce cell damage due to oxidative stress after brain stroke40. We have found that in vitro IGF-I and SCF exert a cooperative neuroprotective effect against oxidative stress, suggesting that they may exert a similar beneficial role in vivo as after brain stroke both factors are upregulated in the lesioned area. Indeed, a cooperative neuroprotective effect of SCF with insulin has been reported41. The intracellular mechanisms mediating cooperation between these two factors involve Erk, a kinase activated by IGF-I.
In summary, cell specific and cooperative actions of IGF-I in brain responses to oxidative challenge underscores the need to design therapeutic strategies that take into account all aspects of biological organization, leading, for example, to cell-specific targeting of anti-aging drugs.
Data availability
figshare: Update 1: Data on the responses of neurons and astrocytes to oxidative injury in the presence of insulin-like growth factor I, http://dx.doi.org/10.6084/m9.figshare.99145670
Author contributions
Laura Genis: designed, analyzed and performed experiments and wrote parts of the manuscript.
David Dávila: designed, analyzed and performed experiments and wrote parts of the manuscript.
Silvia Fernandez: designed, analyzed and performed experiments.
Andrea Pozo-Rodrigálvarez: performed experiments.
Ricardo Martínez-Murillo: performed experiments and contributed materials.
Ignacio Torres-Aleman: designed the study, analyzed the data and wrote the paper.
Competing interests
No competing interests were disclosed.
Grant information
This work was funded by grants of the Spanish Ministry of Science (SAF2010-17036) and Centro Investigacion Biomedica en red Enfermedades Neurodegenerativas (CIBERNED) to IT-A.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We are thankful to L Guinea, M Dominguez and M. Garcia for excellent technical support.
Faculty Opinions recommendedReferences
- 1.
Demaurex N, Scorrano L:
Reactive oxygen species are NOXious for neurons.
Nat Neurosci.
2009; 12(7): 819–820. PubMed Abstract
| Publisher Full Text
- 2.
Doonan R, McElwee JJ, Matthijssens F, et al.:
Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans.
Genes Dev.
2008; 22(23): 3236–3241. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 3.
Yeoman M, Scutt G, Faragher R:
Insights into CNS ageing from animal models of senescence.
Nat Rev Neurosci.
2012; 13(6): 435–445. PubMed Abstract
| Publisher Full Text
- 4.
López-Otín C, Blasco MA, Partridge L, et al.:
The Hallmarks of Aging.
Cell.
2013; 153(6): 1194–1217. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 5.
Braeckman BP, Houthoofd K, Vanfleteren JR:
Insulin-like signaling, metabolism, stress resistance and aging in Caenorhabditis elegans.
Mech Ageing Dev.
2001; 122(7): 673–693. PubMed Abstract
| Publisher Full Text
- 6.
Broughton SJ, Piper MD, Ikeya T, et al.:
Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands.
Proc Natl Acad Sci U S A.
2005; 102(8): 3105–3110. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
Hinkal G, Donehower LA:
How does suppression of IGF-1 signaling by DNA damage affect aging and longevity?
Mech Ageing Dev.
2008; 129(5): 243–253. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 8.
Fernandez AM, Torres-Aleman I:
The many faces of insulin-like peptide signalling in the brain.
Nat Rev Neurosci.
2012; 13(4): 225–239. PubMed Abstract
| Publisher Full Text
- 9.
Guan J, Bennet L, Gluckman PD, et al.:
Insulin-like growth factor-1 and post-ischemic brain injury.
Prog Neurobiol.
2003; 70(6): 443–462. PubMed Abstract
| Publisher Full Text
- 10.
Ryu BR, Ko HW, Jou I, et al.:
Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I.
J Neurobiol.
1999; 39(4): 536–546. PubMed Abstract
| Publisher Full Text
- 11.
Fernandez-Fernandez S, Almeida A, Bolanos JP:
Antioxidant and bioenergetic coupling between neurons and astrocytes.
Biochem J.
2012; 443(1): 3–11. PubMed Abstract
| Publisher Full Text
- 12.
Davila D, Torres-Aleman I:
Neuronal death by oxidative stress involves activation of FOXO3 through a two-arm pathway that activates stress kinases and attenuates insulin-like growth factor I signaling.
Mol Biol Cell.
2008; 19(5): 2014–2025. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
Garcia-Galloway E, Arango C, Pons S, et al.:
Glutamate excitotoxicity attenuates insulin-like growth factor-I prosurvival signaling.
Mol Cell Neurosci.
2003; 24(4): 1027–1037. PubMed Abstract
| Publisher Full Text
- 14.
Finkel T:
Oxidant signals and oxidative stress.
Curr Opin Cell Biol.
2003; 15(2): 247–254. PubMed Abstract
| Publisher Full Text
- 15.
Franco C, Fernandez S, Torres AI:
Frataxin deficiency unveils cell-context dependent actions of insulin-like growth factor I on neurons.
Mol Neurodegener.
2012; 7: 51. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 16.
Pozo-Rodrigalvarez A, Gradillas A, Serrano J, et al.:
New synthesis and promising neuroprotective role in experimental ischemic stroke of ONO-1714.
Eur J Med Chem.
2012; 54: 439–446. PubMed Abstract
| Publisher Full Text
- 17.
Pfaffl MW:
A new mathematical model for relative quantification in real-time RT-PCR.
Nucleic Acids Res.
2001; 29(9): e45. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 18.
Kato H, Faria TN, Stannard B, et al.:
Role of tyrosine kinase activity in signal transduction by the insulin-like growth factor-I (IGF-I) receptor. Characterization of kinase-deficient IGF-I receptors and the action of an IGF-I-mimetic antibody (alpha IR-3).
J Biol Chem.
1993; 268(4): 2655–2661. PubMed Abstract
| Publisher Full Text
- 19.
Lehtinen MK, Yuan Z, Boag PR, et al.:
A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span.
Cell.
2006; 125(5): 987–1001. PubMed Abstract
| Publisher Full Text
- 20.
Papadia S, Soriano FX, Leveille F, et al.:
Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses.
Nat Neurosci.
2008; 11(4): 476–487. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 21.
Dhandapani KM, Wade FM, Wakade C, et al.:
Neuroprotection by stem cell factor in rat cortical neurons involves AKT and NFkappaB.
J Neurochem.
2005; 95(1): 9–19. PubMed Abstract
| Publisher Full Text
- 22.
Chen H, Yoshioka H, Kim GS, et al.:
Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection.
Antioxid Redox Signal.
2011; 14(8): 1505–1517. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 23.
Zhao LR, Singhal S, Duan WM, et al.:
Brain repair by hematopoietic growth factors in a rat model of stroke.
Stroke.
2007; 38(9): 2584–2591. PubMed Abstract
| Publisher Full Text
- 24.
Beilharz EJ, Russo VC, Butler G, et al.:
Co-ordinated and cellular specific induction of the components of the IGF/IGFBP axis in the rat brain following hypoxic-ischemic injury.
Brain Res Mol Brain Res.
1998; 59(2): 119–134. PubMed Abstract
| Publisher Full Text
- 25.
Honda Y, Honda S:
The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans.
FASEB J.
1999; 13(11): 1385–1393. PubMed Abstract
- 26.
Giannakou ME, Partridge L:
Role of insulin-like signalling in Drosophila lifespan.
Trends Biochem Sci.
2007; 32(4): 180–188. PubMed Abstract
| Publisher Full Text
- 27.
Holzenberger M, Dupont J, Ducos B, et al.:
IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice.
Nature.
2003; 421(6919): 182–187. PubMed Abstract
| Publisher Full Text
- 28.
Kappeler L, De Magalhaes Filho CM, Dupont J, et al.:
Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism.
PLoS Biol.
2008; 6(10): e254. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 29.
Nadjar A, Berton O, Guo S, et al.:
IGF-1 signaling reduces neuro-inflammatory response and sensitivity of neurons to MPTP.
Neurobiol Aging.
2008; 30(12): 2021–2030. PubMed Abstract
| Publisher Full Text
- 30.
Grinberg YY, van DW, Kraig RP:
Insulin-like growth factor-1 lowers spreading depression susceptibility and reduces oxidative stress.
J Neurochem.
2012; 122(1): 221–229. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 31.
Gustafsson H, Soderdahl T, Jonsson G, et al.:
Insulin-like growth factor type 1 prevents hyperglycemia-induced uncoupling protein 3 down-regulation and oxidative stress.
J Neurosci Res.
2004; 77(2): 285–291. PubMed Abstract
| Publisher Full Text
- 32.
Heck S, Lezoualc'h F, Engert S, et al.:
Insulin-like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor kappaB.
J Biol Chem.
1999; 274(14): 9828–9835. PubMed Abstract
| Publisher Full Text
- 33.
Offen D, Shtaif B, Hadad D, et al.:
Protective effect of insulin-like-growth-factor-1 against dopamine-induced neurotoxicity in human and rodent neuronal cultures: possible implications for Parkinson's disease.
Neurosci Lett.
2001; 316(3): 129–132. PubMed Abstract
| Publisher Full Text
- 34.
Puche JE, Garcia-Fernandez M, Muntane J, et al.:
Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats.
Endocrinology.
2008; 149(5): 2620–2627. PubMed Abstract
| Publisher Full Text
- 35.
Haigis MC, Yankner BA:
The Aging Stress Response.
Mol Cell.
2010; 40(2): 333–344. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 36.
Beal MF:
Mitochondria, free radicals, and neurodegeneration.
Curr Opin Neurobiol.
1996; 6(5): 661–666. PubMed Abstract
| Publisher Full Text
- 37.
Bishop NA, Lu T, Yankner BA:
Neural mechanisms of ageing and cognitive decline.
Nature.
2010; 464(7288): 529–535. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 38.
Lillig CH, Berndt C:
Glutaredoxins in thiol/disulfide exchange.
Antioxid Redox Signal.
2013; 18(13): 1654–1665. PubMed Abstract
| Publisher Full Text
- 39.
Toth ZE, Leker RR, Shahar T, et al.:
The combination of granulocyte colony-stimulating factor and stem cell factor significantly increases the number of bone marrow-derived endothelial cells in brains of mice following cerebral ischemia.
Blood.
2008; 111(12): 5544–5552. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 40.
Lanfranconi S, Locatelli F, Corti S, et al.:
Growth factors in ischemic stroke.
J Cell Mol Med.
2011; 15(8): 1645–1687. PubMed Abstract
| Publisher Full Text
- 41.
Li JW, Li LL, Chang LL, et al.:
Stem cell factor protects against neuronal apoptosis by activating AKT/ERK in diabetic mice.
Braz J Med Biol Res.
2009; 42(11): 1044–1049. PubMed Abstract
| Publisher Full Text
- 42.
Zheng M, Zhu H, Gong Y, et al.:
Involvement of GMRP1, a novel mediator of Akt pathway, in brain damage after intracerebral hemorrhage.
Int J Clin Exp Pathol.
2013; 6(2): 224–229. PubMed Abstract
| Free Full Text
- 43.
Kukreti H, Amuthavalli K, Harikumar A, et al.:
Muscle-specific microRNA1 (miR1) targets heat shock protein 70 (HSP70) during dexamethasone-mediated atrophy.
J Biol Chem.
2013; 288(9): 6663–6678. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 44.
Wojtalla A, Fischer B, Kotelevets N, et al.:
Targeting the phosphoinositide 3-kinase p110-alpha isoform impairs cell proliferation, survival, and tumor growth in small cell lung cancer.
Clin Cancer Res.
2013; 19(1): 96–105. PubMed Abstract
| Publisher Full Text
- 45.
Cheshenko N, Trepanier JB, Stefanidou M, et al.:
HSV activates Akt to trigger calcium release and promote viral entry: novel candidate target for treatment and suppression.
FASEB J.
2013; 27(7): 2584–2599. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 46.
North AJ, Galazkiewicz B, Byers TJ, et al.:
Complementary distributions of vinculin and dystrophin define two distinct sarcolemma domains in smooth muscle.
J Cell Biol.
1993; 120(5): 1159–1167. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 47.
Vandekerckhove J, Weber K:
Actin amino-acid sequences. Comparison of actins from calf thymus, bovine brain, and SV40-transformed mouse 3T3 cells with rabbit skeletal muscle actin.
Eur J Biochem.
1978; 90(3): 451–462. PubMed Abstract
| Publisher Full Text
- 48.
Drew JS, Moos C, Murphy RA:
Localization of isoactins in isolated smooth muscle thin filaments by double gold immunolabeling.
Am J Physiol.
1991; 260(6 Pt 1): C1332–C1340. PubMed Abstract
- 49.
Lessard JL:
Two monoclonal antibodies to actin: one muscle selective and one generally reactive.
Cell Motil Cytoskeleton.
1988; 10(3): 349–362. PubMed Abstract
| Publisher Full Text
- 50.
Jaarsma D, Teuling E, Haasdijk ED, et al.:
Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice.
J Neurosci.
2008; 28(9): 2075–2088. PubMed Abstract
| Publisher Full Text
- 51.
Shinder GA, Lacourse MC, Minotti S, et al.:
Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis.
J Biol Chem.
2001; 276(16): 12791–12796. PubMed Abstract
| Publisher Full Text
- 52.
Monari M, Cattani O, Serrazanetti GP, et al.:
Effect of exposure to benzo[a]pyrene on SODs, CYP1A1/1A2- and CYP2E1 immunopositive proteins in the blood clam Scapharca inaequivalvis.
Mar Environ Res.
2007; 63(3): 200–218. PubMed Abstract
| Publisher Full Text
- 53.
Shirasawa T, Izumizaki M, Suzuki Y, et al.:
Oxygen affinity of hemoglobin regulates O2 consumption, metabolism, and physical activity.
J Biol Chem.
2003; 278(7): 5035–5043. PubMed Abstract
| Publisher Full Text
- 54.
Katsuki H, Tomita M, Takenaka C, et al.:
Superoxide dismutase activity in organotypic midbrain-striatum co-cultures is associated with resistance of dopaminergic neurons to excitotoxicity.
J Neurochem.
2001; 76(5): 1336–1345. PubMed Abstract
| Publisher Full Text
- 55.
Pedraza-Chaverri J, Maldonado PD, Medina-Campos ON, et al.:
Garlic ameliorates gentamicin nephrotoxicity: relation to antioxidant enzymes.
Free Radic Biol Med.
2000; 29(7): 602–611. PubMed Abstract
| Publisher Full Text
- 56.
Michels S, Trautmann M, Sievers E, et al.:
SRC signaling is crucial in the growth of synovial sarcoma cells.
Cancer Res.
2013; 73(8): 2518–2528. PubMed Abstract
| Publisher Full Text
- 57.
Fei Z, Bera TK, Liu X, et al.:
Ankrd26 gene disruption enhances adipogenesis of mouse embryonic fibroblasts.
J Biol Chem.
2011; 286(31): 27761–27768. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 58.
Wu M, Desai DH, Kakarla SK, et al.:
Acetaminophen prevents aging-associated hyperglycemia in aged rats: effect of aging-associated hyperactivation of p38-MAPK and ERK1/2.
Diabetes Metab Res Rev.
2009; 25(3): 279–286. PubMed Abstract
| Publisher Full Text
- 59.
Abe T, Kaname Y, Hamamoto I, et al.:
Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines.
J Virol.
2007; 81(17): 8953–8966. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 60.
Patrucco E, Notte A, Barberis L, et al.:
PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects.
Cell.
2004; 118(3): 375–387. PubMed Abstract
| Publisher Full Text
- 61.
Fonseca BD, Alain T, Finestone LK, et al.:
Pharmacological and genetic evaluation of proposed roles of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK), extracellular signal-regulated kinase (ERK), and p90(RSK) in the control of mTORC1 protein signaling by phorbol esters.
J Biol Chem.
2011; 286(31): 27111–27122. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 62.
Okkenhaug K, Bilancio A, Farjot G, et al.:
Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice.
Science.
2002; 297(5583): 1031–1034. PubMed Abstract
| Publisher Full Text
- 63.
Chung YW, Kim HK, Kim IY, et al.:
Dual function of protein kinase C (PKC) in 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced manganese superoxide dismutase (MnSOD) expression: activation of CREB and FOXO3a by PKC-alpha phosphorylation and by PKC-mediated inactivation of Akt respectively.
J Biol Chem.
2011; 286(34): 29681–29690. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 64.
Radomska HS, Alberich-Jorda M, Will B, et al.:
Targeting CDK1 promotes FLT3-activated acute myeloid leukemia differentiation through C/EBPalpha.
J Clin Invest.
2012; 122(8): 2955–2966. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 65.
Zhang WH, Wang X, Narayanan M, et al.:
Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death.
Proc Natl Acad Sci U S A.
2003; 100(26): 16012–16017. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 66.
Tsuda M, Toyomitsu E, Komatsu T, et al.:
Fibronectin/integrin system is involved in P2X(4) receptor upregulation in the spinal cord and neuropathic pain after nerve injury.
Glia.
2008; 56(5): 579–585. PubMed Abstract
| Publisher Full Text
- 67.
Ge D, Kong X, Liu W, et al.:
Phosphorylation and nuclear translocation of integrin beta4 induced by a chemical small molecule contribute to apoptosis in vascular endothelial cells.
Apoptosis.
2013; 18(9): 1120–1131. PubMed Abstract
| Publisher Full Text
- 68.
Wu YS, Lu HL, Huang X, et al.:
Diabetes-induced loss of gastric ICC accompanied by up-regulation of natriuretic peptide signaling pathways in STZ-induced diabetic mice.
Peptides.
2013; 40: 104–111. PubMed Abstract
| Publisher Full Text
- 69.
Raucci F, Di Fiore MM:
Localization of c-kit and stem cell factor (SCF) in ovarian follicular epithelium of a lizard, Podarcis s. sicula.
Acta Histochem.
2011; 113(6): 647–655. PubMed Abstract
| Publisher Full Text
- 70.
Laura Genis, David Dávila, Silvia F, et al.:
Data on the responses of neurons and astrocytes to oxidative injury in the presence of insulin-like growth factor I. figshare. 2014. Data Source
In response to points raised by the first reviewer Marta Margeta:
1: Why are neurons cultured under depolarizing ... Continue reading We would like to respond to some of the points raised by the reviewers.
In response to points raised by the first reviewer Marta Margeta:
1: Why are neurons cultured under depolarizing conditions (25 mM KCl)?
As shown in http://cshprotocols.cshlp.org/content/2008/12/pdb.rec11550 cerebellar granule cells require 25 mM KCl to survive in vitro. We checked ourselves a long time ago this specific requirement and confirmed that without it these neurons do not survive.
4: Neuronal viability is established by counting “all GFP-positive cells”; the authors therefore need to show that their neuronal cultures are at least 95% and preferably 99% pure (i.e., do not contain a significant population of GFP-positive glial cells).
We established neuronal-enriched culture methods long ago using neurofilament (Fig 1A in Torres-Aleman et al., Neuroscience, 1998) or beta3 tubulin as neuronal marker (Garcia-Galloway et al., Mol. Cell Neurosci., 2003). We have 90-95% cells showing neuronal markers. Less than 5% stain for glial markers. Cell morphology also helps to avoid counting non-neuronal GFP cells.
In response to points raised by the third reviewer Vince Russo:
1: In figure 1B, IGF-I ELISA, the conditioned media from astrocytes and neuronal cells is analysed for IGF-I levels but the values are express in ng/ug of protein, why?
IGF-I levels are expressed as ng/µg protein because supernatants were concentrated by lyophilization and re-suspended in ELISA buffer. Protein in supernatants was measured by Bradford to normalize IGF-I levels.
In response to points raised by the first reviewer Marta Margeta:
1: Why are neurons cultured under depolarizing conditions (25 mM KCl)?
As shown in http://cshprotocols.cshlp.org/content/2008/12/pdb.rec11550 cerebellar granule cells require 25 mM KCl to survive in vitro. We checked ourselves a long time ago this specific requirement and confirmed that without it these neurons do not survive.
4: Neuronal viability is established by counting “all GFP-positive cells”; the authors therefore need to show that their neuronal cultures are at least 95% and preferably 99% pure (i.e., do not contain a significant population of GFP-positive glial cells).
We established neuronal-enriched culture methods long ago using neurofilament (Fig 1A in Torres-Aleman et al., Neuroscience, 1998) or beta3 tubulin as neuronal marker (Garcia-Galloway et al., Mol. Cell Neurosci., 2003). We have 90-95% cells showing neuronal markers. Less than 5% stain for glial markers. Cell morphology also helps to avoid counting non-neuronal GFP cells.
In response to points raised by the third reviewer Vince Russo:
1: In figure 1B, IGF-I ELISA, the conditioned media from astrocytes and neuronal cells is analysed for IGF-I levels but the values are express in ng/ug of protein, why?
IGF-I levels are expressed as ng/µg protein because supernatants were concentrated by lyophilization and re-suspended in ELISA buffer. Protein in supernatants was measured by Bradford to normalize IGF-I levels.