Keywords
animal model, complement, lupus nephritis, systemic lupus erythematosus, therapy
This article is included in the Lupus nephritis and neuropsychiatric lupus collection.
animal model, complement, lupus nephritis, systemic lupus erythematosus, therapy
The involvement of the complement system in the pathogenesis of a number of autoimmune diseases including SLE is well accepted, yet its exact role is still not clear. On one hand, hereditary deficiencies of early classical pathway complement components predispose patients to SLE. On the other hand, activation of complement by immune complexes (ICs) is certainly a prominent feature in SLE that promotes tissue injury. Therefore, an imbalance of the complement system in either direction and the respective roles of the classical and alternative pathways can have complex effects on the disease phenotype. The current review will discuss the dual roles of complement in SLE, and in particular in lupus nephritis (LN).
The complement system is an important part of innate immunity which defends the host against infectious microorganisms, clears ICs and dead cells, and serves as a bridge between innate and adaptive immunity1. Complement can be activated through classical, alternative and mannose-binding lectin (MBL) pathways, each with different initiators (shown schematically in Figure 1).
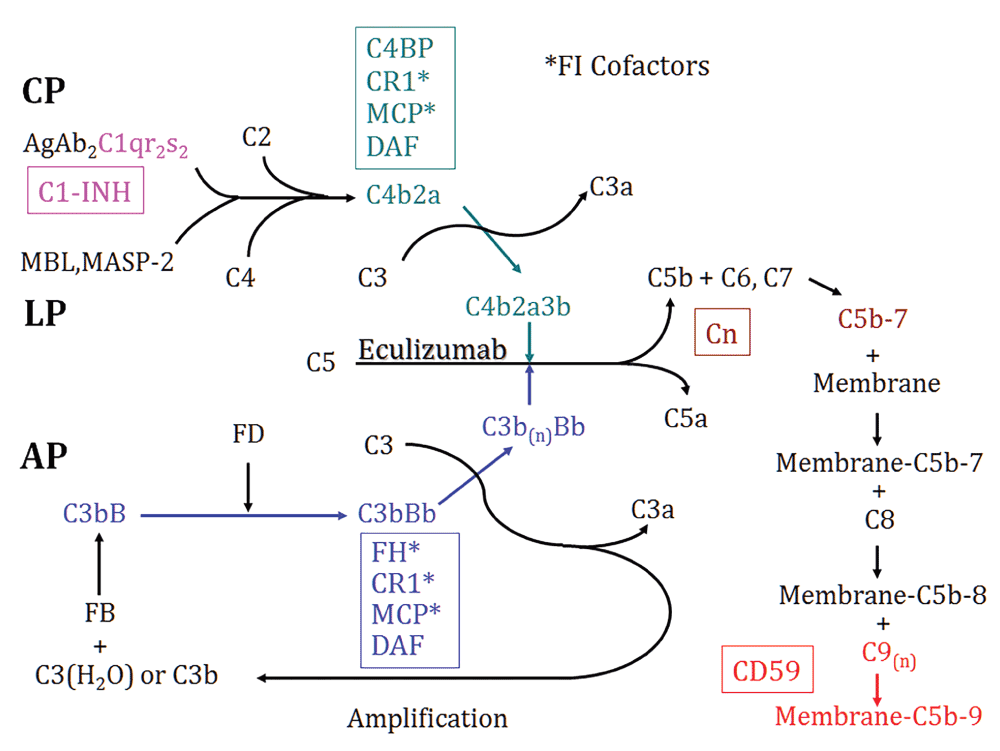
Shown are the three activation pathways – classical (CP), MBL (LP) and alternative pathways (AP) and the common intermediates of activation, C3 and C5 convertases. Regulatory proteins are in boxes, matched by color to the sites of inhibition. Anti-C5 antibody (Eculizumab) is also depicted. Cn, Clusterin; FI, complement factor I.
The classical pathway is activated when the C1q subunit of C1 binds with high avidity to the Fc portion of immunoglobulin (Ig) M (CH3 domain) or IgG (CH2 domain) in ICs. Besides Igs, C1q also binds and facilitates the removal of apoptotic cells, which affords it an important role in immune tolerance. Loss of complement self-regulatory protein CD46 (also known as Membrane Cofactor Protein, MCP), on apoptotic cells (by shredding into microparticles or cluster into blebs) was suggested as the mechanism of driving C3 opsonization and promotion of efferent removal of apoptotic cells2. The binding of C1q to its target triggers its conformational change and self-activation of C1r, followed by the activation of C1s3. Activated C1 (as the multiprotein C1qr2s2 complex) cleaves both C4 and C2 to generate C4a/C4b and C2a/C2b. C4b2a acts as a C3 convertase which cleaves and activates C3.
The alternative pathway is spontaneously activated by a C3 conformation change, which occurs slowly but continuously to generate hydrolyzed C3 (C3(H2O)). C3(H2O) then binds complement factor B (CFB) in the fluid phase. Upon factor B’s cleavage by complement factor D (CFD, also known as adipsin), an initial C3 convertase C3(H2O)Bb is formed, which can be stabilized by properdin, and promotes its own production to form C3bBb, the alternative pathway C3 convertase1.
The binding of MBL to terminal carbohydrate groups on certain microbes leads to the activation of the MBL-associated serine proteases (MASPs). Activated MASP cleaves C4 to C4a and C4b. Immobilized C4b induces the binding of C2 which is also cleaved by MASP and generates the C4b2a C3 convertase4.
Irrespective of the pathway of activation, cleavage of C3 and C5 ultimately occurs, with the generation of the C3a and C5a anaphylatoxins, C3b opsonins, and C5b to start the non-enzymatic assembly of the C5b-9 membrane attack complex, which can result in cellular death or activation after membrane insertion5, which also occurs in erythrocytes (E) in paroxysmal nocturnal hemoglobinuria (PNH)6.
Given the potency of the complement system, natural fluid-phase and cell membrane-bound regulatory proteins acting throughout the three cascades are essential to prevent activation and injury to host tissues (Figure 1)1. The regulators of complement activation (RCA) gene family on human chromosome 1q3.2 (and a comparable location in mouse chromosome 1) includes MCP, complement receptor 1 (CR1), decay accelerating factor (DAF, also known as CD55), C4b-binding protein (C4bp), and CFH7,8. These proteins inhibit complement activation through interactions of their conserved short consensus repeats (SCRs) with fragments of C3 and/or C49.
MCP is a cell surface glycoprotein which serves as a cofactor for factor I-mediated cleavage of C3b and C4b, thereby inhibiting the formation of C3/C5 convertases10. More recently, it was found that besides acting as a complement inhibitor, MCP also regulates T cell subsets during an immune response by promoting the activation of TH1 cells and their IL-10 production11. Studying of MCP in rodents has its obstacles, due to its restricted expression predominantly in testicular germ cells12, while in humans MCP has been identified in all cell types except erythrocytes10. DAF is a glycosylphosphatidylinositol (GPI)-anchored membrane protein, which binds C3b and C4b and accelerates the decay of C3 (C4b2a in the classical pathway and C3bBb in the alternative pathway) and C5 (C4b2a3b in the classical pathway and C3bBb3b in the alternative pathway) convertases13. CR1 is a single chain transmembrane glycoprotein which has the combined functions of DAF and MCP: it accelerates the decay of C3 and C5 convertases like DAF and it also serves as a cofactor for complement factor I-mediated inactivation of C3b and C4b into iC3b and iC4b with a similar function to MCP14. The plasma proteins, C4bp and complement factor H (CFH) also have cofactor activity for factor I-mediated inactivation of plasma C4b and C3b, in the classical and alternative pathways, respectively. Specific to rodents is the 65-kDa rodent complement regulatory protein, more commonly referred to as CR1-related gene/protein y (Crry) which has combined decay-accelerating and factor I cofactor activity for C3b and C4b, similar to CR115. Lastly, at either “end” of the complement cascades are C1-inhibitor (C1-INH) or CD59, which inhibit C1 activation and C5b-9 formation, respectively16,17.
SLE is an autoimmune disorder caused by loss of tolerance to self-antigens, the production of autoantibodies, and deposition of complement-fixing ICs in injured tissues. SLE is characterized by a wide range of clinical manifestations and targeted organs, with LN being one of the most serious complications18. The pathogenic roles of complement activation in human SLE were indicated from years of clinical observations: low total complement hemolytic activity (CH50) and decreased C3 and C4 levels have been found in about 75% of SLE patients with focal nephritis and 90% in patients with diffuse nephritis19. Additionally, the co-localization of Ig isotypes IgG, IgA, and IgM with C1q, C4 and C3 (and C5b-9) (the so called “full house” pattern) in the glomeruli is almost exclusively present in glomeruli of patients with lupus nephritis20. Finally, complement split products such as C3d and C5b-9 can also be detected in the urine of SLE patients21.
While frequent mutations in CFH and MCP found in atypical hemolytic uremic syndrome (aHUS) are not associated with lupus nephritis, SLE patients carrying these mutations do have an earlier onset of lupus nephritis22. SLE patients with lymphopenia and neutropenia were found to have lower level of DAF and CD59 on neutrophils, and SLE patients with anemia had lower expression of CD59 and CR1 on erythrocytes, and the expression is correlated with disease activity23. In neutrophils from SLE patients, IC down-regulates the levels of CR1 transcripts, both directly, and indirectly via inhibiting interferon (IFN) -γ induced CR1 expression, an example of how lupus activity affects CR1 expression24. Nonetheless, higher CR1 expression on SLE patient leucocytes indicates a better prognosis25, while lower leucocyte MCP expression is associated with lupus remission26. These findings not only suggest the pivotal roles of the complement system in the pathogenesis of SLE but may also potentially provide noninvasive markers for the disease activity.
Impaired IC handling plays an important role in the pathogenesis in LN. Since the complement system is required at all steps of normal IC metabolism, any number of alterations can lead to pathological glomerular IC accumulation, particularly in conditions of IC excess, as in SLE. Studies have shown the association of SLE with low levels of CR1 on erythrocytes, a key site of binding and transferring IC to the mononuclear phagocyte system27, suggesting that a defective erythrocyte/IC-clearing system may be related to SLE pathogenesis.
In contrast to the widespread belief that generation of complement activation products in kidney and other disease sites is proinflammatory, patients with homozygous deficiencies of the C1 proteins (C1q or C1r/s) or C4 have a high prevalence (> 80%) of autoantibodies and SLE-like disease28. Successful treatment of C1q-deficient SLE patients with fresh frozen plasma or hematopoietic stem cell transplantation was also reported29,30. It is also noteworthy that in SLE patients, anti-C1q antibodies are associated with proliferative lupus nephritis, and anti-C1q antibody levels may indicate renal disease activity31. More recently, it was reported that in the presence of sera from individuals deficient in C1q, C4, C2 or C3, phagocytosis of apoptotic cells was decreased compared with studies using normal sera, indicating important roles of all these complement classical pathway components in clearance of apoptotic and necrotic cells32. Circulating cell-derived microparticles in patients with SLE carry more IgG, IgM, and C1q, and IgG-bearing microparticles are associated with autoantibodies and complement activation33. Interestingly, in the very rare cases of homozygous deficiency of C3, the most critical protein that affects all three pathways of complement activation, there is no association with SLE34.
Complement also plays important roles in thrombotic complications associated with SLE. The risk of thrombosis is particularly high in SLE patients with antiphospholipid (aPL) autoantibodies. One explanation is that aPL-containing ICs bind to platelets which can subsequently activate the classical complement pathway, as supported by the fact that SLE patients have increased levels of C1q, C3d and C4d on their platelets, especially in patients with history of venous thrombosis. Sera from SLE patients with aPL autoantibodies has a higher capacity to activate the classical pathway on heterologous platelets35. Though C1q and C4d deposition on platelets is not specific for SLE, this is associated with venous thrombosis36. One study also suggests that small dense HDL particles may activate complement system and is related to subclinical atherosclerosis in SLE patients37.
Two of the best studied murine models that spontaneously develop lupus-like syndromes are the F1 cross between New Zealand Black and New Zealand White mice (NZB/W) and the MRL/MpJ-Tnfrsf6lpr/lpr/J (MRL/lpr) strain38. Similar to the female predominance in humans, only female NZB/W mice develop SLE. Both models have B cell hyperactivity, autoantibodies, hypocomplementemia, circulating and glomerular-bound ICs, and severe nephritis. As in humans, there is plenty of circumstantial evidence that complement activation is actively involved in the pathogenesis of glomerular disease in these mice. In early stages (4 and 5 months of age in MRL/lpr and NZB/W mice, respectively), granular deposition of mouse IgG, IgA, IgM and C3 are present largely in the mesangium, coincident with histopathology showing mesangial proliferation. As the disease progresses, there are glomerular capillary wall IC deposits, proliferation of intrinsic endothelial and mesangial cells, and infiltration with inflammatory cells. Eventually, crescent formation (more often in MRL/lpr mice) and glomerulosclerosis (more often in NZB/W mice) occurs, and mice die of renal failure38.
The manipulation of individual complement proteins through genetic techniques in lupus mouse strains and functional inhibition through the use of specific antibodies or antagonists have provided considerable insight into how complement is involved in this disease. Given that C3 is the common point connecting all three pathways in complement activation and is tightly regulated naturally, many of the studies in lupus mice have concentrated on activators and regulators of C3.
DAF is a ubiquitously expressed GPI-anchored membrane protein that inhibits C3 activation through all pathways by inhibiting formation and accelerating decay of the C3/C5 convertase13. The relevance of DAF to lupus is suggested by the fact that DAF-deficient MRL/lpr mice had exacerbated lymphoproliferation, anti-chromatin autoantibody production and dermatitis, particularly evident in females, while nephritis appeared to be unaffected39. Furthermore, DAF-deficient MRL/lpr mice also deficient in C3 developed comparable lymphadenopathy, splenomegaly and anti-chromatin autoantibodies to what is seen in the complement sufficient mice, suggesting the protective effect of DAF in MRL/lpr autoimmunity is complement-independent. DAF-sufficient MRL/lpr chimeras with DAF-deficient MRL/lpr bone marrow developed significantly attenuated dermatitis compared with that in DAF-deficient MRL/lpr chimeras with DAF-sufficient MRL/lpr bone marrow, indicating that the protective effect of DAF on dermatitis is attributable to local expression40. The exacerbated dermatitis can be explained by the fact that DAF is strongly expressed in the skin while Crry is not.
Like human CR1, Crry is an intrinsic membrane complement inhibitor that inhibits C3 convertases of all pathways, combining activities of human DAF and MCP15. Given that the Crry-deficient mice generated by Molina et al. have complete embryonic lethality from unrestricted maternal complement activation41, which makes generation of a straightforward Crry-deficient murine lupus model impossible. The critical role of Crry in regulating complement in the kidney came from our study of transplantation of Crry−/−C3−/− kidneys into wildtype hosts. Due to lack of Crry, there was marked inflammation in the tubulointerstitium which led to complete failure of the transplanted kidney within weeks (while the appropriate controls, including wild type kidneys transplanted into wild type or Crry−/−C3−/− mice remained normal)42. On the other hand, transgenic mice developed by our group that overexpressed a soluble form of Crry had less severe lupus nephritis as determined by blood urea nitrogen (BUN) and albuminuria measurements when crossed into the MRL/lpr strain. Since the spontaneous mortality in lupus mice is largely due to kidney disease, this translated into prolonged survival, while the underlying abnormal autoimmunity was not affected43. To make this complement inhibition more applicable to human SLE treatment, a recombinant soluble form of Crry (Crry-Ig) was also used in MRL/lpr mice. In chronic usage from early in the autoimmune disease until the end-stage, inhibited complement activation by Crry-Ig ameliorated lupus nephritis44. Interestingly, transcript profiling experiments showed that excessive matrix components such as collagens I, III, IV and VI were overexpressed in control MRL/lpr mice, which could be suppressed by complement inhibition with Crry-Ig. Potential explanations for these phenomena include Crry-Ig-mediated reductions in connective tissue growth factor and TGF-β1 expression, suggesting these profibrotic mediators are downstream of complement-induced injury and contribute to the progressive glomerulosclerosis in MRL/lpr mice45.
In both human and experimental SLE, CR1/CR2 expression decreases over time, suggesting that this plays a role in disease46,47. Yet, MRL/lpr mice deficient in CR1/CR2 had significantly lower levels of total IgG3 and specific IgG3 rheumatoid factor, supporting the role of CR1/CR2 in production of IgG3 in response to autoantigens. Nonetheless, this decrease of IgG3 autoantibodies did not lead to a reduction in features of lupus nephritis48.
C4bp is a major soluble complement inhibitor of the classical pathway. Elevated serum levels of C4bp in SLE patients suggested its involvement in this disease49. More detailed studies addressing this possibility were performed by the Braun group, using a C4bp deficient MRL/lpr mouse model. While serum C4 levels are lower in MRL/lpr mice compared with normal C57BL/6 mice, starting as early as 3 weeks of age (i.e., prior to autoimmune disease onset), surprisingly deficiency of C4bp affected neither serum C4 levels nor the classical pathway hemolytic activity. With an unaffected complement classical pathway, C4bp deficiency did not lead to a significantly different outcome in these mice, which had similar amounts of glomerular C3 and IgG deposition, glomerulonephritis, tubulointerstitial inflammation, proteinuria and renal function. Moreover, the systemic immune response was not affected by the absence of C4bp in these mice50. One explanation for the negligible role of C4bp in lupus development in MRL/lpr mice is that the extensively activated classical pathway in this model simply overwhelms the absence of C4bp. In this situation, “a gain of function” of C4bp may reveal more information, as purified human C4bp successfully inhibited the classical complement pathway, leading to delayed or reduced disease development in these models51.
In physiological situations, there is spontaneous continuous low-level alternative pathway activation that is restrained by effective complement regulation. While the traditional thinking is that SLE is induced through IC-directed classical pathway activation, the involvement of the alternative pathway has also been suggested in many studies. Gilkeson et al. demonstrated that CFB- or CFD-deficient MRL/lpr mice had reduced glomerular C3 deposition associated with less severe glomerular histopathology52,53. These results imply that IC-directed classical pathway activation can recruit the potent alternative pathway to further amplify generation of C3 and C5 activation products. Complement factor H-related (CFHR) protein C is absent in the circulation of MRL/lpr and NZB/W mice before and after the onset of lupus, implying that polymorphic variation may contribute to the development of SLE54. Our group demonstrated CFH-deficient MRL/lpr mice developed a severe inflammatory diffuse lupus nephritis by 12 weeks, characterized by glomerular hyalinosis (“wire-loops”), and messagial, endocapillary and extracapillary proliferation, that resulted in the death of 50% of animals by 13 weeks of age55. These findings indicate that in contrast with the spontaneous disease in older CFH-deficient mice on mixed backgrounds56, loss of CFH accelerates the development of lupus nephritis and recapitulates the functional and structural features of human disease in MRL/lpr mice.
C5b-9 deposition was found in diseased lupus kidneys more than two decades ago. Yet, compared with extensive studies focusing on C3 activation and its regulation, fewer studies have been done to investigate the downstream events following C3 activation in the pathogenesis of SLE. Wang et al. used a specific monoclonal antibody to inhibit C5 function in NZB/W mice. Six months of continuous therapy led to significantly delayed onset of proteinuria, improved renal pathological changes and prolonged survival, implicating a role of products of the terminal complement pathway, C5a and C5b-9 in lupus nephritis57. On the other hand, CD59a-deficient MRL/lpr mice had exacerbated skin disease and lymphoproliferation, through a complement-independent, autoimmunity regulatory mechanism, which is conferred by CD59a’s expression on both bone marrow-derived cell and peripheral tissues58.
The C3a and C5a anaphylatoxins are generated through complement activation when C3 and C5 are activated and cleaved. Increased expression of C3aR expression was found in glomeruli in human lupus nephritis59. C3aR and C5aR expression were significantly up-regulated at both the mRNA and protein levels and accompanied by a wider cellular distribution in MRL/lpr mouse kidneys60,61. This upregulated expression starts before the onset of kidney disease, supporting the idea that they may be involved in the development of disease, rather than simply being a consequence. Chronic administration of a specific C3aR antagonist (SB290157) led to significantly reduced kidney disease and prolonged survival in MRL/lpr mice61. Similarly, when C5a signaling was blocked in our studies with a specific antagonist60 or in those by Braun et al. through gene targeting62, MRL/lpr mice animals displayed attenuated renal disease and prolonged viability. The effects of blocking C3aR and C5aR in lupus mice had certain features in common, including less renal neutrophil and macrophage infiltration, apoptosis, and IL-1β expression60,61. Effects on chemokine expression were distinct, with C3aR- and C5aR-inhibited MRL/lpr mice having reduced CCL5 (RANTES) and CXCL2 (MIP-2) expression, respectively. C3aR-inhibited mice also had increased phosphorylation of protein kinase B (Akt), which we considered suggestive that C3aR signals promote renal cell apoptosis through an Akt pathway61. In C5aR-deficient MRL/lpr mice, there was a reduction in CD4+ T cell renal infiltration, lower titers of anti-double stranded DNA antibodies, and inhibition of interleukin (IL)-12 p20 and IFN-γ production, suggesting that Th1 responses are important to link C5a signaling in lupus nephritis62. In contrast, C3aR-deficient MRL/lpr mice had elevated autoantibody titers, more glomerular crescents and more severe intrarenal vasculitis, though it did not affect long-term renal injury or survival63. The mechanisms of different outcomes between short term (by using an antagonist) and long-term (by using gene-targeting) blockade of C3aR signaling in this lupus model still need to be clarified. This may be due to the fact that the particular C3aR antagonist (SB290157) used in this study also has partial agonist activity64.
Since C3 is the converging point for all three complement pathways, alterations in C3 activation through manipulating its regulators, such as Crry or DAF, or blockade of the effects of C3 activation with inhibitors of C3aR, C5aR, or C5 have shown C3 activation is an important factor in the development of SLE. Surprisingly, C3 deficiency does not affect the development of nephritis in MRL/lpr mice, while glomerular IgG deposition is significantly increased65. This study is consistent with the important role of complement, and in particular C3, in the clearance of ICs66. The finding that homozygous deficiency of early components of the classical pathway other than C3 predispose to SLE suggests that physiological function of these molecules are protective in SLE. While the exact mechanism is still unknown, studies found that mice with generated C1q and C4 deficiencies had impaired ability to clear apoptotic debris67, leading to the accumulation of potentially immunogenic autoantigens and initiation of an autoimmune reaction in the right genetic setting. C1q-deficient mice had increased mortality and higher titers of autoantibodies, with 25% of the mice developing glomerulonephritis, characterized by glomerular IC deposits and apoptotic debris68. Further work suggests that C1q and DNase1 are important in the degradation of chromatin derived from necrotic cells69. It was also found that the IgG autoantibodies were responsible for the inhibition of macrophage removal of apoptotic cells through C3 deposition in MRL/lpr and NZB/W mice70. On the other hand, C3 deposition on nucleic acids is significantly reduced when dsDNA-specific IgG autoantibodies are present in the serum, which increase as disease progresses in MRL/lpr mice71.
In both human and experimental models of lupus nephritis, the complement system has a dual role. The classic pathway contributes to its protective role in the clearance of apoptotic material and circulating IC32,72. In theory chronic systemic complement inhibition may increase circulating IC and exacerbate disease44. To selectively target the desired complement regulator to the site of tissue injury, Tomlinson et al. used CR2 as the “guide” of complement inhibitors, given that sites of complement activation are “marked” by the presence of C3d73, and CR2 has natural affinity for C3d. Thus, low doses of chimeric CR2-DAF and CR2-CD59 efficiently protected target cells from complement attack74. CR2-DAF was targeted to the glomerulus in lupus nephritis while soluble DAF failed74. Long term (8 week) treatment of diseased MRL/lpr mice with low doses of CR2-Crry and CR2-CFH provided significant complement inhibition locally in the lupus glomerulus, which conferred significant reduction of proteinuria, as well as prolonged survival in these mice75,76. CR2-CFH treatment also significantly reduced glomerulonephritis76. The fact that selective alternative pathway inhibition by CR2-CFH provided significant reduction in glomerulonephritis while total complement inhibition with CR2-Crry did not, support the hypothesis that the alternative and classical pathways of complement have distinct roles in the pathogenesis of lupus nephritis76,77.
Investigations of complement activation in lupus nephritis did not only suggest potential therapies, but also diagnostic and monitoring tools. Using CR2-targeted MRI, glomerular C3b/iC3b/C3d deposition in MRL/lpr mice was found to increase with disease activity, suggesting its usage as a marker of onset and severity of lupus glomerulonephritis, which could be used to monitor the disease course and its response to therapy, repeatedly and noninvasively78.
Strategies to manipulate the complement system in different human diseases have followed from successful animal studies, including those using recombinant intrinsic complement regulators and blocking antibodies. In addition to the treatment approaches indicated in studies using experimental models discussed above, we will focus on several promising therapeutic approaches that have been used in the treatment of human diseases, and may potentially extend to the treatment of human SLE and lupus nephritis.
Soluble CR1 (sCR1) was first developed in 1990, and has both decay accelerator and cofactor activity in classical and alternative complement pathways. A current therapeutic form of soluble CR1 designated as TP10 (Avant Immunotherapeutics, Nedham, MA) has been used in clinical trials in several human diseases, including acute respiratory distress syndrome79, ischemia-reperfusion injury in patients undergoing lung transplantation80 or cardiac surgery81. These studies showed that sCR1 was well tolerated, with low development of anti-sCR1 antibodies81. More recently, soluble CR1 was found to prevent dysregulation of the alternative pathway C3 convertases in dense deposit disease (DDD) and C3 glomerulonephritis (C3GN), supporting clinical trial of soluble CR1 as a treatment for DDD and C3GN82.
C1 esterase inhibitor (C1-INH) has been marketed as replacement therapy for hereditary angioedema (HAE) and ischemia-reperfusion injury. Plasma derived C1-INH has shown to be effective in prophylaxis, reducing the frequency of attacks of HAE, and shortening the duration of the acute attacks83. Recombinant human C1-INH also proved to be effective in the treatment of HAE patients during acute attacks84. C1-INH was also reported to successfully ameliorate both complement deposition on red blood cells (RBCs) and hemolysis in autoimmune hemolytic anemia85.
The most extensively used antibody targeting the complement system is a humanized monoclonal antibody (Eculizumab, Alexion Pharmaceuticals, Inc., Cheshire, CT) that directly binds human C5 and prevents its cleavage to C5a and C5b. In March 2007, Eculizumab (Soliris) is the first and the only US Food and Drug Administration (FDA) approved complement-inhibitor used in the treatment of human diseases. It was approved by the FDA for the treatment of Paroxysmal Nocturnal Hemoglobinuria (PNH), an acquired disorder of GPI-linked proteins including DAF and CD59 characterized by spontaneous complement activation and C5b-9-mediated hemolysis. It was subsequently approved for the treatment of atypical hemolytic uremic syndrome (aHUS) in 2011. Different phase 3 clinical trials showed that Eculizumab treatment showed better stabilization of hemoglobin, and reduced intravascular hemolysis with significant clinical improvements86, even with long term treatment87. Eculizumab increases aHUS patients’ platelet count, with 80% of patients achieving thrombotic microangiopathic event-free status88. With this beneficial effect of Eculizumab in aHUS, it is encouraging that a phase I trial has shown it is safe, with a prolonged interval of complement inhibition in SLE patients. Moving to the kidney, a multi-center phase II trial in the United States in which 122 patients with idiopathic membranous nephropathy were enrolled in a randomized placebo-controlled study of Eculizumab has been completed. Unfortunately, there was no difference comparing treatment with placebo in the primary outcome variable of urinary protein excretion. Because of the short-term treatment strategy (16 weeks) in a long-term disease, the study design may have been insufficient to uncover a true therapeutic effect. This is supported by the finding of an apparent benefit in patients enrolled in an open-label extension. Based on the impressive effects of long-term treatment with anti-C5 effects in lupus nephritis in NZB/W lupus mice57, Furie et al. conducted a phase I study with eculizumab in 24 patients with SLE. In this single center, randomized, placebo-controlled, double-blind, dose-ranging trial, patients were given a single intravenous dose of Eculizumab or placebo and followed for two months. Only mild adverse events were reported, and complement inhibition for 10 days in the 8 mg/kg group was observed89. We also designed a multi-center phase II trial using Eculizumab in proliferative lupus nephritis supported by the United States’ National Institutes of Health. Unfortunately, after enrollment of our first patient, this study encountered logistical delays and ultimately came to a complete halt. While this reflects the difficulties in clinical trials for a disorder such as lupus nephritis, based on what is known about the pathophysiology we remain interested in the potential efficacy of this drug in the treatment of lupus nephritis.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)