Keywords
Bayesian Networks, Protein Interaction Networks, Saccharomyces Cerevisiae, Signaling Pathways
This article is included in the Bioinformatics gateway.
Bayesian Networks, Protein Interaction Networks, Saccharomyces Cerevisiae, Signaling Pathways
For biologists and scientists in the life sciences, the successful sequencing of the genome is only one step out of many involved in understanding organisms. This has produced a lot of information that will not be useful unless refined. Biologists are interested in understanding the intricacies of the workings of the cells of an organism – the activities and reactions of such an organism to its environment. This information is useful in designing necessary interventions in order to modify the biological mechanisms of an organism or its reactions to external stimuli.
According to the central dogma of molecular biology, genes are composed of DNA which is transcribed into RNA and the RNA is then translated into protein. Ultimately, all organisms are composed mainly of proteins in different forms and quantity.
Proteomic data and protein-protein interaction data from organisms form a key component in understanding an organism due to the major role played by proteins in cellular mechanisms. Protein-protein interactions are the foundation of biological mechanisms such as signal transduction, cell cycle control, DNA replication and transcription and enzyme-mediated metabolism1,2.
As a result of these interactions, understanding of organisms is facilitated by modeling the Protein Interaction Network (PIN) with a network constructed using the protein-protein interaction data. With a model such as this, a lot can be learned of the organism from its reaction to external stimuli and the effects of interventions on the biological mechanisms of the organism. For instance, it has been shown that the phenotypic effects of the deletion of a single gene depend on the position of that gene in the complex web of protein interactions3.
Apart from the importance of the protein-protein interactions map in studying the machinery of the proteome and the cellular behaviour of an organism, they are also practically important in the creation of interventions aimed at producing desired phenotypic outcomes such as new drug designs or disease prevention4,5.
Protein-protein interaction data from organisms are obtained on a large scale using a number of high throughput techniques such as Yeast Two-Hybrid (Y2H), Co-Immunoprecipitation (Co-IP), Mass Spectrometry etc. These high throughput techniques have however, been identified to have high rates of false positives and false negatives. False positive interactions are protein-protein interactions that are reported to exist with any of the experimental techniques but do not exist in reality, while false negative interactions are true interactions that do not get reported using an experimental technique. Rates of false positives in protein interaction data have been reported to be as high as 50%6–8. As a result of analysis based on the integration of gene expression level measurement data and protein-protein interaction data, only about 30–50% of the interactions have been suggested to be biologically relevant. Reference 9 reported 47% true protein-protein interactions where a Paralogous Verification Method (PVM) was applied. The PVM may have performed better owing to its incorporation of information on paralogs of other organism to strengthen the biological evidence.
These high rates of protein-protein interaction data inaccuracy are due to peculiarities of the techniques used to generate them. For instance, unlike other affinity-based methods that cannot detect transient interacting proteins, Tandem Affinity Purification (TAP-tag) tag methods can detect transient interacting proteins which are however lost during the purification process10.
Furthermore, these new high throughput methods of detecting protein interactions have no doubt rapidly generated much more data than have been collected by traditional methods in small scale experiments. This thus makes it impractical to start verifying each of these interactions by the traditional methods used in small scale experiments11.
In order to make sense of the vast data and obtain insightful information, these data need to be subjected to analytical procedures that will extract signal from the noise. This task of analyzing genomic data takes a computational approach due to the magnitude of the information involved. In reducing this level of noise in the protein interaction data different computational techniques aimed at improving the reliability of the data are applied. To predict true interactions between protein pairs, many authors have suggested a number of methods for estimating and assigning reliabilities to the interactions in the experimental data. These methods include using a logistic regression distribution function over a number of parameters to assign confidence scores to the interactions17,18, the use of expression profile and paralogs to assign reliability scores to already observed interactions9 the use of maximum likelihood technique for the estimation of domain-domain interactions in order to infer protein-protein interactions7. For computational biologists, the challenge would be the development of methods of transforming the high-throughput data obtained from these different sources into biological insights.
In this paper, we seek to bridge the gap between protein-protein interaction data and other biological data in constructing useful signaling pathway models that will lead to insightful knowledge of biological processes. We propose a probabilistic approach using Bayesian networks to assign weights to protein-protein interactions. These weighted interactions are then used to construct the weighted PIN from which signaling pathways are predicted. Refer to Figure 1 for the schematic of the computational approach used. This work thus describes a computational means to clean up the background noise inherent in the various methods of proteomic data acquisition in order to better understand bio-molecular mechanisms.
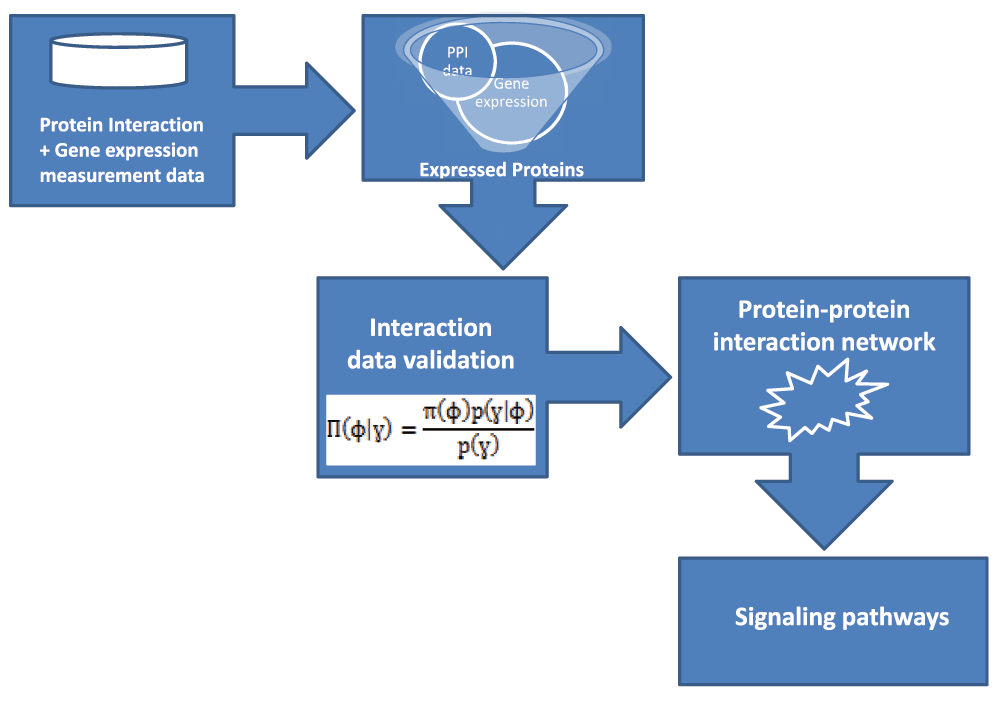
Protein interaction data was obtained from the publicly available Saccharomyces Genome Database (SGD). The protein interaction data is an amalgamation of the interactions obtained using eight different high throughput experimental procedures - Yeast Two-Hybrid, Affinity Capture Mass Spectrometry, CO-Purification, Affinity Capture Western, Biochemical Activity, Reconstituted Complex, Protein-Peptide and Far Western. This data contained 22,650 interactions between 2554 different proteins.
The S. cerevisiae expression measurement data was obtained from the Yeast Cell Cycle Analysis Project of the Stanford University. The data is housed at a publicly available database maintained by the Saccharomyces Genome Database at the Department of Genetics, School of medicine. The Yeast Cell Cycle Analysis project aimed at identifying all the genes whose mRNA levels are regulated by the cell cycle12. This data is available at the Yeast Cell Cycle Analysis Project site. The data contained the expression profiles of 800 proteins of the S. cerevisiae organism.
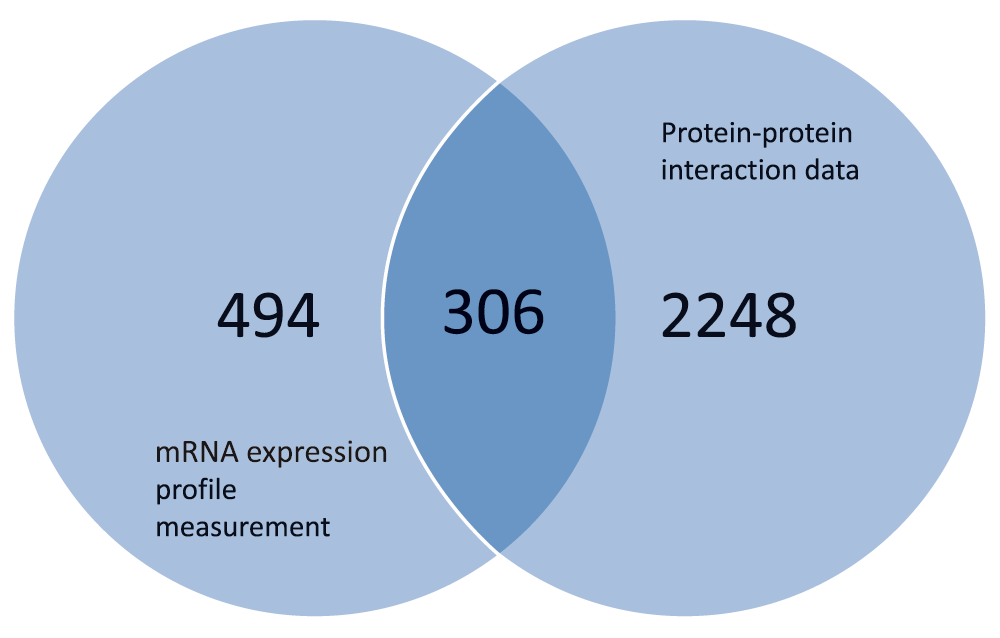
The processing of the data obtained from the Yeast protein-protein interaction data and the Yeast Expression measurement was carried out by first filtering for the proteins that have expression level measurement. Only the proteins in the protein-protein interaction dataset that were also present in the gene expression measurement data were used. This was done based on the hypothesis that proteins occurring in the same complex and are known to physically interact have higher correlation than proteins that are not known to directly interact. This hypothesis is supported by13–16, where it has been observed that true protein interactions have a high mRNA expression for the proteins involved. The filtration of the dataset produced 306 protein-protein interactions that have expression level measurements from the 22,650 protein interactions and the 800 gene expression measurements. These 306 protein-protein interactions represent the intersection of the two datasets as depicted in Figure 2. With reference to the yeast protein interaction data and expression measurement data respectively, these figures correspond to 0.013% and 0.382% of the original dataset respectively.
In this work, the probability estimation of protein interactions was done using a Bayesian probabilistic model. According to Bayes' theorem, the posterior probability density is proportional to the prior probability density and the likelihood function.
Our interest is in drawing inference about the parameter ϕ from a probability model p(ɣ|ϕ) to give rise to observed data ɣ. Allocating a prior probability π(ϕ) to the parameters assuming they are uncertain, we can obtain a posterior probability according to Bayes' theorem where p(ɣ) which is the marginal density for ɣ is obtained by integrating over the prior. Refer to Equation 1.
Equation (1) can be rewritten as equation (2) since π(ϕ|ɣ) is a function of ϕ for observed ɣ which shows the direct proportionality between the posterior probability and the product of the likelihood and the prior probability19.
In order to make use of the Bayesian model, there must be an approximation or full specification of the prior probability distribution and the likelihood function. The first step in the determination of the likelihood function which is based on the probability of observing the data is to fix a probability distribution f(θ) where θ is the parameter defining the probability distribution.
In outcome space, for a given dataset (Y1, Y2, Y3, … ,Yn), the probability of observing the dataset given θ is written as
In parameter space, the likelihood function in terms of the probability of observing the dataset given θ is
For a Bernoulli distribution, the probability distribution is
Therefore for a sample of N observations (Y1, Y2, Y3, …,Yn), the joint distribution is as Equation 6, and can be rewritten as Equation 7
The likelihood function determines what value of θ makes the dataset (Y1, Y2, Y3, …,Yn) most probable.
Estimating the maximum likelihood of the parameter θ, we maximize the function with respect to θ and then set it to zero to obtain the Maximum Likelihood Estimation of the parameter θ20.
We formalize the problem of constructing a weighted graph that is instrumental in building a PIN. Let (V, E, w) be the protein-protein interaction network where V = p0, p1,...pn is the set of all proteins and E = {e = (pi, pj) | pi, pj ϵ V} is the set of interactions among these proteins in the set V, and w is the weight of each edge that belongs to E. w being the weight of the interaction between two proteins (pi, pj) is a measure of the reliability of interaction between the two proteins (pi, pj) obtained using the Bayesian probabilistic approach described above.
The vertices of the interaction graph are contained in the set of the unique proteins obtained after the computation of the reliability of interaction between the protein pairs i.e. |V| = 306, and the edges of the graph are the set of interactions between these proteins in V.
The protein-protein interaction graph is constructed with an undirected sparse graph due to the sparse nature of biological networks.
The implementation of the algorithm for this computational technique was done in Java programming language using the Java Universal Network Graphics (JUNG) framework for graphs. The JUNG framework is an open-source collection of libraries providing common language for modeling, analyzing and visualizing any data that can be represented as a graph or network. The JUNG framework is extensible in order to tailor it to specific needs and also includes implementation of a host of algorithms for network analysis, graph theory and data mining.
The graph implementation in JUNG supports the representation of the different types of graphs such as directed and undirected graph, multimodal graphs, graphs with parallel edges and hypergraphs.
For this work, we used the JUNG 2.0.1 API released in January, 2010 which can be found at http://jung.sourceforge.net.
A pathway is an ordered list of distinct proteins in V such that each consecutive pair is found in E21.
Given an undirected sparse graph G = (V, E, w) and a pair of nodes {(pi, pj) | (pi, pj) ϵ V} corresponding to the starting and ending proteins respectively, we wish to find a simple path from pi to pj which will be a segment of a pathway.
With the graph constructed, which is the PIN of the S. cerevisiae organism based on the data supplied, we queried the graph with a pair of proteins (pi, pj) ϵ V which are respectively the starting protein and ending proteins of the path of interest. We are interested in having a simple path corresponding to the signal transduction path from the starting protein to the ending protein returned by the search algorithm.
The search is done using a Depth First Search (DFS) algorithm. The start protein becomes the root node for the algorithm and examines all the outgoing nodes to it, expands the first child node of the apparent tree and progressively continues the search until the target node (the ending protein) is found. If the DFS algorithm however encounters a node that has no children, it backtracks to the previous node to continue exploring the children nodes.
In order to understand and make meaning of the pathway segments that are obtained from the PIN, we compared the proteins to their functional annotation. Mapping proteins in known signaling pathways and PINs to their functional annotations has an important function. The proteins in an organism may have similar biological functions such that one protein effectively replaces another in a pathway, then such proteins should share the same set of gene annotation terms. The Gene Ontology annotation, which is a functional annotation scheme, provides this basis for the identification of functional description of proteins and their interactions with other proteins and other molecules.
In this work, we used the Gene Ontology (GO) annotations to interpret the pathway segments that have been identified from the protein-protein interaction network constructed for the S. cerevisiae organism.
To validate the protein-protein interaction data that we used in this work, we applied the method that was described in section II-B to first filter the data. As was described the filtration of the data was done by integrating the gene expression measurement of the regulated Yeast Cell-cycle in order to obtain a dataset that is an intersection of both datasets.
This step was taken based on the hypothesis that there is a high correlation between the expression levels of truly interacting proteins13–16,22 and also using the gene expression measurement as a source of biological information23–25 for the computational inference.
Using the protein-protein interaction data comprising 22,650 interactions between 2554 unique proteins and the gene expression levels of 800 genes, we applied the computational approach based on Bayesian probability described earlier. Further in the validation process, the application of the Bayesian probabilistic model on the data to estimate the posterior probability of an interaction existing between two proteins given the biological evidence produced the weight estimate for the interactions. With the estimation of the interaction weight and the rejection of interaction weights below the threshold obtained from the mean expression level measurements, we obtained a dataset containing 306 protein-protein binary interactions. This dataset was used in constructing the PIN of the S. cerevisiae organism.
The 306 protein-pairs represented the proteins that had expression profile measurement, which corresponds to the intersection set of the two datasets. With reference to the protein-protein interaction data and the gene expression level measurement, this is a mere 0.013% and 0.382% of the original dataset respectively.
We applied the method described in section 2.7 to identify pathway segments in the constructed PIN for S. cerevisiae. Given a graph G = (V, E), a pathway has been described as an ordered list of distinct proteins in V such that each consecutive pair is found in E21. With a starting protein and an ending protein of interest, a simple path between these two corresponds to a pathway. Due to the size limitation of the expression measurement dataset used and the effective reduction in the overall number of proteins used to construct the graph, we were only able to identify pathway segments. A pathway segment is a chain of interacting proteins which is a part of a larger pathway. Some of the resulting pathways identified with this technique are presented in Table 1 and Table 2. These tables elucidate the protein description, the GO function and GO process of the proteins involved in the pathway segments as obtained from the AmiGO website http://www.geneontologyproject.org/go.
| Protein | Gene | Protein Information | Gene Ontology Function | Gene Ontology Process |
|---|---|---|---|---|
| YMR163C | Inp2 | Peroxisome-specific receptor important for peroxisome inheritance; co-fractionates with peroxisome membranes and co-localizes with peroxisomes in vivo; physically interacts with the myosin V motor Myo2p; INP2 is not an essential gene29 |
Myosin binding
Interacting selectively and non-covalently with any part of a myosin complex; myosins are any of a superfamily of molecular motor proteins that bind to actin and use the energy of ATP hydrolysis to generate force and movement along actin filaments. [GO:0017022] |
Peroxisome inheritance
The acquisition of peroxisomes by daughter cells from the mother cell after replication. In Saccharomyces cerevisiae, the number of peroxisomes cells is fairly constant; a subset of the organelles are targeted and segregated to the bud in a highly ordered, vectorial process. Efficient segregation of peroxisomes from mother to bud is dependent on the actin cytoskeleton, and active movement of peroxisomes along actin filaments is driven by the class V myosin motor protein, Myo2p. [GO:0045033] |
| YOR326W | MYO2, CDC66 | Type V myosin motor involved in actin-based transport of cargos; required for the polarized delivery of secretory vesicles, the vacuole, late Golgi elements, peroxisomes, and the mitotic spindle; MYO2 has a paralog, MYO4, that arose from the whole genome duplication30 | Actin filament binding Interacting selectively and non- covalently with an actin filament, also known as F-actin, a helical filamentous polymer of globular G-actin subunits. [GO:0051015] | Cell division The process resulting in the physical partitioning and separation of a cell into daughter cells. Source: GOC: go_curators Comment Note that this term differs from ‘cytokinesis ; GO:0000910’ in that cytokinesis does not include nuclear division. [GO:0051301] |
| YCL063W | VAC17, YCL062W | Phosphoprotein involved in vacuole inheritance; degraded in late M phase of the cell cycle; acts as a vacuole-specific receptor for myosin Myo2p31,32 |
Protein anchor
Interacting selectively and non-covalently with both a protein or protein complex and a membrane, in order to maintain the localization of the protein at a specific location on the membrane. [GO: 0043495] | No Information Available |
| YER150W | SPI1 | GPI-anchored cell wall protein involved in weak acid resistance; basal expression requires Msn2p/Msn4p; expression is induced under conditions of stress and during the diauxic shift; SPI1 has a paralog, SED1, that arose from the whole genome duplication33 |
Molecular_function
Elemental activities, such as catalysis or binding, describing the actions of a gene product at the molecular level. A given gene product may exhibit one or more molecular functions. [GO: 0003674] |
Response to acid
Any process that results in a change in state or activity of a cell or an organism (in terms of movement, secretion, enzyme production, gene expression, etc.) as a result of an acid stimulus. The acid may be in gaseous, liquid or solid form. [GO: 0001101] |
| Protein | Gene | Protein Information | Gene Ontology Function | Gene Ontology Process |
|---|---|---|---|---|
| YIL026C | IRR1, SCC3 | Subunit of the cohesin complex; which is required for sister chromatid cohesion during mitosis and meiosis and interacts with centromeres and chromosome arms; relocalizes to the cytosol in response to hypoxia; essential for viability34,35. |
Chromatin Binding
Interacting selectively and non-covalently with chromatin, the network of fibers of DNA, protein, and sometimes RNA, that make up the chromosomes of the eukaryotic nucleus during interphase. [GO: 0003682] |
fungal-type cell wall organization
A process that is carried out at the cellular level which results in the assembly, arrangement of constituent parts, or disassembly of the fungal- type cell wall. [GO: 0031505] |
| YIL126W | STH1p, NPS1 | ATPase component of the RSC chromatin remodeling complex; required for expression of early meiotic genes; essential helicase- related protein homologous to Snf2p36,37. |
DNA-dependent ATPase activity Catalysis of the reaction: ATP + H2O = ADP + phosphate; this reaction requires the presence of single- or double-stranded DNA, and it drives another reaction. [GO: 0008094] |
ATP-dependent chromatin remodeling Dynamic structural changes to eukaryotic chromatin that require energy from the hydrolysis of ATP, ranging from local changes necessary for transcriptional regulation to global changes necessary for chromosome segregation, mediated by ATP- dependent chromatin-remodelling factors. [GO: 0043044] |
| YDL003W | MCD1 | Essential alpha-kleisin subunit of the cohesin complex; required for sister chromatid cohesion in mitosis and meiosis; apoptosis induces cleavage and translocation of a C-terminal fragment to mitochondria; expression peaks in S phase38,39. |
Chromatin binding
Interacting selectively and non-covalently with chromatin, the network of fibers of DNA, protein, and sometimes RNA, that make up the chromosomes of the eukaryotic nucleus during interphase. [GO: 0003682] |
Establishment of mitotic sister chromatid cohesion The process in which the sister chromatids of a replicated chromosome become joined along the entire length of the chromosome during S phase during a mitotic cell cycle. [GO:0034087] |
| YJL074C | SMC3 | Subunit of the multiprotein cohesin complex required for sister chromatid cohesion in mitotic cells; also required, with Rec8p, for cohesion and recombination during meiosis; phylogenetically conserved SMC chromosomal ATPase family member40. |
ATPase activity
Definition Catalysis of the reaction: ATP + H2O = ADP + phosphate + 2 H+. May or may not be coupled to another reaction. [GO: 0016887] |
Meiotic sister chromatid cohesion
The cell cycle process in which sister chromatids of a replicated chromosome are joined along the entire length of the chromosome during meiosis. [GO: 0051177] |
Table 1 and Table 2 present some of the pathway segments identified using the computational approach proposed in this paper. The understanding of the paths is facilitated by using Gene Ontology associations to understand the biological processes the proteins are involved in. A signaling pathway is characterized by a starting protein that is a receptor at the membrane and ends with a transcription factor.
From Table 1 we identified a pathway segment {YMR163C- YOR326W- YCL063W- YER150W} along with the genes coding for each of the proteins using GO annotation.
The pathway segment starts with the protein YMR163C, identified to be a receptor important for peroxisome inheritance. Signaling pathways are often characterized by an activator at the membrane of the cell binding to a receptor to initiate the chain of signal transduction. These peroxisomes are organelles that metabolize fatty acids and are numerous in the S. cerevisiae organism. By blocking peroxisome transport through point mutants in the MYO2p gene that binds to it, the levels of MYO2p gene expression increased26. The implication of this is that signal is transmitted to the mother cell to stop further peroxisome transfer by lowering INP2 gene expression. The next protein, YOR326W, in the pathway segment is coded for by the MYO2p gene whose level of expression is modified in the activation of signal that is relayed to alter the level of the INP2 gene that codes for the YMR163C receptor protein. The next protein in the chain, YCL063W, coded for by the gene VAC17 has been identified to be a vacuole-specific receptor for myosin MYO2P and is involved in vacuole inheritance, a molecular anchoring function. The last protein in the pathway segment, YER150W, coded for by the gene SPI1 contributes to transcriptional regulation induced under conditions of stress during the diauxic shift27.
It is observed that this pathway which signals the start of the process of meiosis suddenly breaks off to a gene (SPI1) that participates in catalysis at the molecular level. This is not abnormal as these are pathway segments and not the full transduction pathway activated by the receptor protein.
In a similar approach, 28 used protein-protein interaction data and expression data to model pathways. They ranked candidate signaling pathways of interacting proteins using expression data based on the rationale that proteins in the same signaling network must simultaneously exist with the activation of the pathway; the genes coding for these proteins must also under the same environmental factors required for the signaling network and about the same time, be transcribed.
Their approach to predicting pathways included specifying the starting protein, a membrane protein, and an ending protein of interest, such as a DNA-binding protein, based on a prior knowledge of genetic relationship between them. In their findings, the pathways that the algorithm identified were not complete pathways owing to incomplete maps.
21 also applied a computational approach that is similar to our own by assigning scores to protein-protein interaction data, creating a PPI network from the data and mining signaling pathways from the network. The parameters for the search on the network included a starting protein and an ending protein as well as the length of the pathway segment. Although their approach involved training the algorithm using association rules mining from known pathways, they were only able to mine pathway segments too.
The incompleteness of pathways mined from using computational techniques on protein-protein interaction data can be attributed to false negative interactions that were not detected by the high throughput experiments that generated the data.
Furthermore, a number of computational techniques that have been applied to cleaning the noise in the protein-protein interaction data used often entails eliminating some data presumed to be noise from the dataset. The proteins removed in this manner could be important proteins that would then be missing in the modeled PIN. Our own approach involved filtering the protein-protein interaction data with the gene expression measurement data such that only the proteins with expression level measurement were used in the construction of the protein interaction network. This resulted in a reduction of the 22,650 pair-wise interactions by the gene expression measurement for just 800 proteins to 306 pair-wise interactions. This reduction in the size of the data used to construct the protein interaction network was a constraint on the number of pathways identified using this approach.
In this paper, we proposed a simple computational approach to identify signaling pathways in PINs by first estimating true interactions within protein-protein interaction data obtained from high throughput experimental techniques which are susceptible to generating high rates of false positive and false negative interactions. We proposed a technique using Bayesian Probability to estimate the probability of true interactions between two proteins and assigned weights to the pair wise interaction based on this. Using the validated protein-protein interaction data, we constructed a PIN of the S. cerevisiae organism from where simple paths between two proteins of interest were mined. Using the Gene Ontology annotation to understand the biological process taking place within the pathway, we were able to identify a pathway which signals the start of the process of meiosis, albeit broken off for want of more data.
Knowledge of signaling pathways are generally useful in designing biological interventions on an organism aimed at producing specific desired outcomes such as new drugs design and disease prevention and control.
F1000Research: Dataset 1. Yeast Expression Data, 10.5256/f1000research.7591.d11032541
F1000Research: Dataset 2. Protein-protein interaction data, 10.5256/f1000research.7591.d11032642
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)