Keywords
Ebolavirus, pathogenicity, transmission, Major histocompatibility complex (MHC), Immune epitopes, protein interactions
This article is included in the Emerging Diseases and Outbreaks gateway.
This article is included in the Ebola Virus collection.
Ebolavirus, pathogenicity, transmission, Major histocompatibility complex (MHC), Immune epitopes, protein interactions
Ebolavirus is responsible for explosive though rare outbreaks of haemorrhagic fever common in equatorial Africa. The virus is transmitted from person to person through infected body fluids such as faeces, vomit and blood (Dowell et al., 1999). Ebolavirus comprises mainly of four subtypes; Zaire, Sudan, Cote d’Ivoire and Reston. Ebola ancestors are not clearly defined, though Marburg virus, which belongs in the same family as Ebola (Filoviridae; a grouping consisting of nonsegmented, negative-stranded RNA viruses) is a possible ancestor (Mayo & Pringle, 1998; Sanchez et al., 1993).
The average rate of case fatalities due to Ebolavirus is 78% (Kuhn, 2008). The Ebola outbreak in 2014 was considered the largest in history and estimates show about 2,473 infections with 1350 deaths occurring (WHO). The 2014 outbreak was first identified in February in Guinea, West Africa (Burke, 2003), then in March it spread into Liberia, to Sierra Leone in May and finally to Nigeria towards the end of July. This outbreak was considered the largest because of its exponential expansion, doubling every 34.8 days. In major cities such as Conakry (Guinea), Monrovia (Liberia), Freetown (Sierra Leone) and Lagos (Nigeria) where viral emergence was observed, the spectre of local and international viral dissemination was raised.
Currently, no approved drugs have been identified against Ebolavirus disease, though rapid diagnosis, intensive care and support is playing a major role in improving survival. Characteristics of the Ebola disease include; high fever, headache, body aches, intense weakness, stomach pain, and lack of appetite. Manifestations of Ebola in patients are: vomiting, diarrhea, rash, impaired kidney and liver function and for critical cases, external and internal bleeding (http://www.nih.gov/news/health/aug2014/od-29.htm).
The current Ebola epidemic raised awareness worldwide given its international transmission observed in the US and Spain. Several genetic approaches to understanding the disease are currently being tested, the majority investigating the genomic composition of the virus, pathogenic pathways within the host and immune variants (MHC-HLA class I & II) commonly expressed by Ebola disease survivors and non-survivors (Sullivan et al., 2003). The HIV, ENV glycoprotein (gp160) and Ebola virus envelope glycoprotein (VGP) are genetically closely related hence, on disrupting the glycoprotein transmembrane domain of MHC class I at the cell surface, a facilitated viral evasion of the immune system was observed (Ploegh, 1998). This viral interaction at the cell surface is considered to occur with the CD209 antigen (DC-SIGN), making the VGP a prominent area of research for drug/vaccine development.
The Ebolavirus genome is 19 kb in length, with seven open reading frames that encode structural proteins including virion envelope glycoprotein (VGP), nucleoprotein (NP), and matrix proteins VP24 and VP40; nonstructural proteins, including VP30 and VP35; and the viral polymerase (Sanchez et al., 2001). Unlike the Marburg virus, the GP open reading frame of Ebolavirus has two gene products, a soluble 60- to 70-kDa protein (sGP) and a full-length 150- to 170-kDa protein (GP) that inserts into the viral membrane (Sanchez et al., 1996; Volchkov et al., 1995), through transcriptional editing.
The adaptive immune and inflammatory systems both respond to viral infection synchronously, such that some types of cells (e.g.: monocytes and macrophages) become principal targets for disease pathogenesis. This infection criteria was first suggested based on observations of Ebolavirus immunohistochemical localization in vivo, therefore, mononuclear phagocytes (monocytes and macrophages), endothelial cells, and hepatocytes constitute the main infectious targets (Baskerville et al., 1978; Baskerville et al., 1985). The recognition of specific Ebolavirus VGP epitopes by monoclonal antibodies conveyed immune protection in a murine model (Wilson et al., 2000) and in guinea pigs (Parren et al., 2002) infected with Ebolavirus.
From the current information known about viral interaction with host genes and pathways favoring cytokine activation, this study was undertaken to analyze the relationship between predicted viral antigenic epitopes and their population frequency as a means of transmission risk. This Ebola species-epitope variation was used to decipher viral species diversity and their related pathway, and how this interplay favors disease intensity within the host. The different viral epitopes identified in this study and their related immune peptides served as a means for predicting which viral species could be commonly found in a given Ebolavirus epidemic. This study opens up epidemiological aspects of viral pathogenesis and possible targets for drug/vaccine design.
The sequence alignment for all retrieved protein variants of different Ebola sequences showed a 1.3% identity across all sequences and a 14.8% pairwise identity across amino acid residues for all sites. These values further improved for each genomic variant analysed for all Ebola species as follows: 23.3% identity across all sites and 68.8% pairwise identity for nuclear proteins, 17.5% identity across all sites and 57.2% pairwise identity for virion glycoproteins and 23.9% identity across all sites and 70.6% pairwise identity respectively for matrix proteins. The phylogenetic analysis showed two main clusters (Figure 1A) of nuclear proteins on one arm (blue cluster) and the other arm contained glycoproteins in a sub cluster (red), and matrix proteins in another sub cluster (blue). Mammalian related immune protein sequences associated with viral antigenicity composed the rest of the sequences. The red sub cluster described above was further subjected to phylogenetic analysis and four sub clusters were identified (Figure 1B) with the red composed of the Zaire Ebolavirus (29 variants), the black composed of the Reston Ebolavirus (14 variants) the green composed of the Sudan Ebola virus (11 variants) and the blue composed of Bundibugyo and Taï forest Ebola viruses (8 variants as indicated by the name) (Table 1). The Marburg virus separated independently and appears to be the ancestral strain from which Ebolaviruses diverged. The host-virus phylogenetic clustering based on related functional sequence features associated with viral genomic structures.
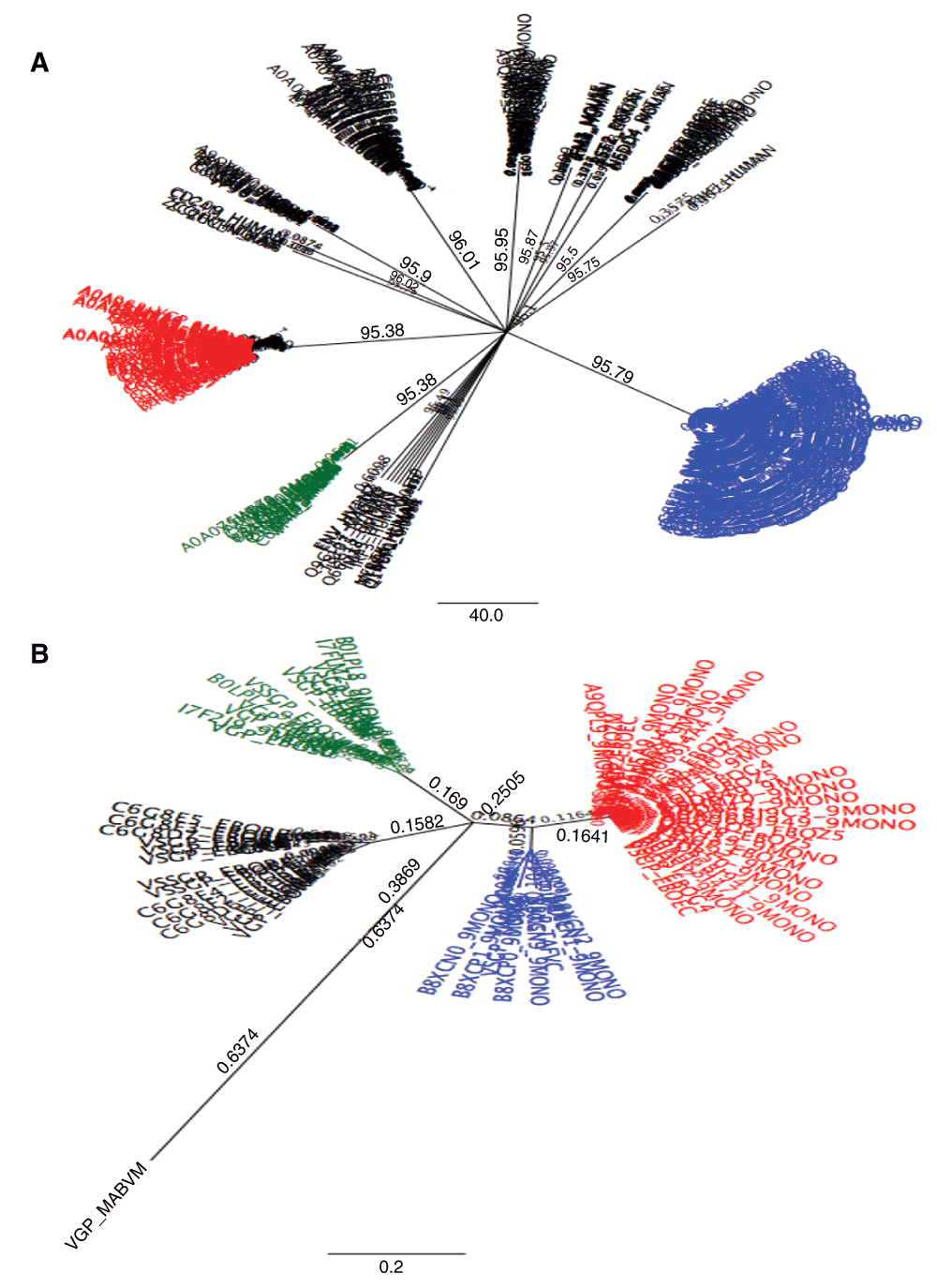
348 amino acid sequences for Ebolavirus species and mammals were aligned and their phylogeny on the unrooted tree showed two distinct clades (1A) wherein the blue cluster belonged to nuclear protein sequences of the viral species while the other cluster contained viral glycoprotein sequences (red sub cluster) and viral matrix proteins (green sub cluster) and the rest were dominantly mammalian proteins. 1B) The viral glycoproteins were subjected to further phylogenetic analysis and the unrooted tree showed four main clusters belonging to different species as follows; the red composed of Zaire Ebolavirus (29 variants), the black composed of the Reston Ebolavirus (14 variants) the green composed of the Sudan Ebolavirus (11 variants) and the blue composed of Bundibugyo and Taï forest Ebolaviruses (8 variants).
29 variants were identified for Zaire Ebolavirus, 14 variants for Reston Ebolavirus, 11 for Sudan Ebolavirus, 3 variants for Bundibugyo Ebolavirus (B) and 5 variants for Taï forest Ebolavirus (T).
The predicted network showed 69 nodes with 123 edges (Figure 2). The network analysis distinguished from most important to least important genes (nodes) depicted by name size (Figure 2). The three most important nodes in descending order included: GRB2 (Growth factor receptor bound protein 2), IKKE (Inhibitor of nuclear factor kappa-B Kinase subunit epsilon) and UBIQ/UBB (Polyubiquitin-B). Plotting neighborhood connectivity (number of neighbors per node) against clustering coefficient (CC) [ratio of connected edges N for neighbors of node n compared to the maximum number of edges M for node n (N/M)] (Figure 3), showed that several nodes (n = 16) did not cluster but had several neighbors. The largest cluster had a coefficient of 0.15 to 0.30. The node distribution based on the best-fit line (4.346 = 8.858x) showed a correlation value of r2 = 0.319 indicating skewedness towards most connected nodes. Based on all GO molecular functions derived from the network, focus was laid on those genes involved with immune and virus related functions which were; LCK (a tyrosine-protein kinase) and CD209 (a pathogen recognition receptor) respectively. LCK associated functions included: protein kinase activity (GO 4672, P-value = 1.9585E-9) (Figure 5B), interspecies interaction between organisms (GO 44419, P-value = 1.3293E-7), T cell activation (GO 42110, P-value = 1.1774E-4), receptor binding (GO 5102, P-value = 7.5709E-3), SH2 domain binding (GO 42169, P-value = 1.2270E-2), CD8 receptor binding (GO 42610, P-value = 1.5663E-2) and CD4 receptor binding (GO 42609, P-value = 2.5648E-2). CD209 associated functions included; Intracellular virion transport (GO 46795, P-value = 2.5648E-2), endocytosis (GO 6897 P-value = 1.1185E-2), signal transducer (GO: 4871, P-value = 1.1671E-2), peptide antigen transport (GO 46968, P-value = 3.4042E-2) and peptide binding (GO 42277, P-value = 4.4976E-2). The protein kinase pathway associated with LCK, showed two clusters of interacting proteins (Figure 4A) with the largest (9 genes) containing LCK while that for peptide binding associated with CD209, showed non-interacting proteins (Figure 4B). Three proteins, Bone marrow stromal antigen (BST2), Serine/threonine-protein kinase (TBK1) and Interferon regulatory factor 3 (IRF3), had as function, response to virus (GO 9615, P-value = 2.4498E-2) and of the three, TBK1 was identified in the protein kinase pathway. Two viral related proteins identified on the network were ENV [envelope glycoprotein gp160 for the HIV-1 virus which binds to CD4 cells (GO 19031, P-value = 4.0529E-2)] and VP40 (virus matrix protein, a structural protein most commonly found in Ebolavirus and promotes virus assembly and budding) though both showed no protein interactions. Identification of regulatory elements targeting the network showed that, 66 drugs have been developed as targets for several genes within the network, of which proto-oncogene tyrosine-protein kinase (src) and LCK ranked first and second respectively. A few transcription factors like; signal transducer and activator of transcription 1-alpha/beta (STAT1), T-cell leukemia homeobox protein 1 (TLX1), transcription factor PU.1 (SPI1), cyclin dependent kinase inhibitor 1A, and cyclin-dependent kinase inhibitor 1A (CDKN1A), regulated genes including nuclear factor NF-κ-B p105 subunit (NFKB1), p100 subunit (NFKB2), CD209 and SRC respectively (Figure 5D). Several microRNA molecules (n = 1399) targeting several genes within the network, mapped to different disease pathways with the most important being hsa05131 (pathogenic Escherichia coli infection) and hsa01430 (cell communication) while major pathway gene related domains were; IPR001664 (intermediate filament protein) and IPR000108 (neutrophil cytosol factor 2).
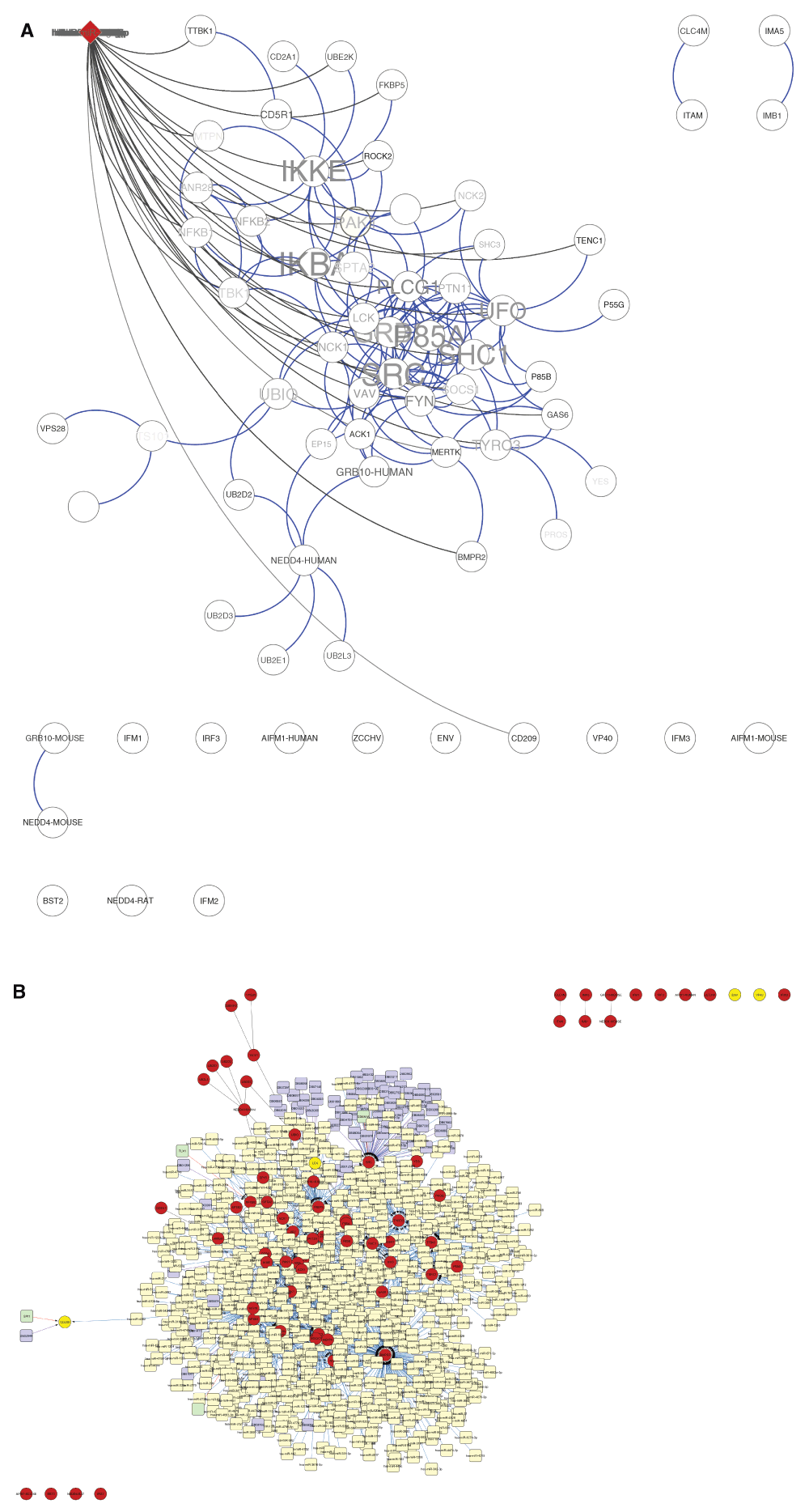
A) The 348 extracted sequences were used to predict a protein interaction map with 69 nodes and 123 edges. Several mammalian sequences captured on the network showed direct interactions with each other with one main protein (LCK) a tyrosine kinase was involved with virus interactions and signalling functions. Name size related to number of interactions each protein had within the network. Two viral proteins were captured in the network; VP40 a matrix protein and ENV a glycoprotein. The red node at top of the network interacting with several other nodes represents a cluster of microRNAs within the network and their related genes they regulate based on their interactions. B) Several regulatory elements were seen to interact with the nodes of interest (red circles). MiRNAs (yellow squares), drug targets (purple squares), transcription factors (light green squares).
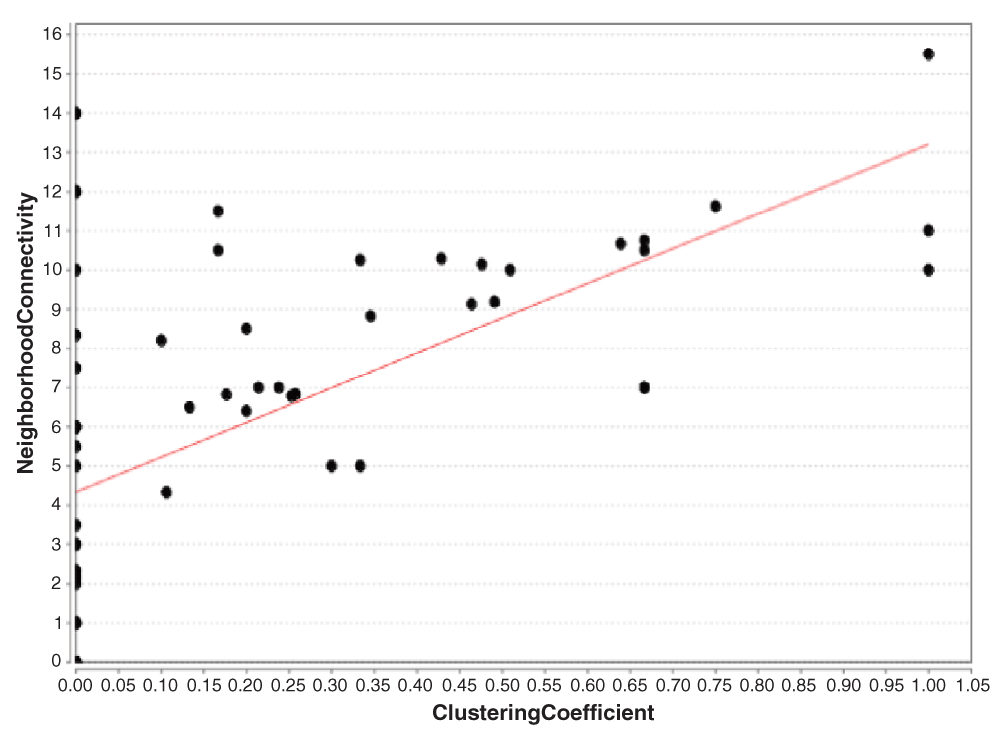
This graph shows that several genes had neighbors up to 14 proteins but no clusters were formed due to no interactions. For those that clustered, majority were between 0.15 and 0.30 defining a high zone of interacting genes within cells.

A) for the several pathways identified with LCK, protein kinase pathway showed three clusters (blue nodes) with LCK involved in the largest cluster (9 genes). B) The peptide binding cluster (brown nodes) involving CD209 a pathogen recognition receptor gene showed no direct interactions between each other.
Antigenic epitopes were predicted for MHC class I and MHC class II. Four (A) against three (B) epitopes for VCP and MXP respectively were validated for MHC class I alleles, 221 (C) against 154 (D) epitopes for VGP and MXP respectively were validated for MHC class II HLA-DRB1 alleles and 41 (E) against 11 (F) epitopes for VGP and MXP respectively were validated for MHC class II HLA-DQA-B alleles. Generally their peptide antigenicity was higher that host population frequency.
The most antigenic epitopes for each virus structural proteins (glycoproteins and matrix proteins) were statistically evaluated for their peptide antigenic score against peptide frequency (count) for the HLA allele types (Figure 5A–F). For HLA type I, the VGP epitope peptides showed t = 0.24338 while the MXP proteins showed t = 0.09190. For the HLA type II (HLA-DRB alleles), the VGP showed t = 2.8506E-107 while the MXP showed t = 2.21965E-49 and for HLA type II (HLA-DQA-A alleles), the VGP showed t = 8.96323E-08 while the MXP showed t = 0,046921205. For HLA class I alleles, four main epitopes (FLLQLNETI, FLTNTIRGV, FQVDHLTYV and NMTNFLFQV) were predicted for VGP while three main epitopes (AIMLASYTV, ALLTGSYTI and LTMVITPDY) were predicted for MXP. Both viral structural proteins showed high frequency values for HLA*A02:01 and HLA*A02:02 alleles (Figure 6A, Table S2) with VGP higher for the latter than the former and vice versa for MXP. The validated epitopes for VGP showed that; FLLQLNETI was specific to HLA*A02:02 while FQVDHLTYV and NMTNFLFQV was variably distributed for all the selected HLA class I alleles (Figure 6B). The validated epitopes for MXP showed that; AIMLASYTV was variably distributed for selected HLA class I alleles while ALLTGSYTI and LTMVITPDY were allele specific (Figure 6C). FLLQLNETI was specific to Zaire Ebolavirus with a unique occurrence while the other epitopes were either specific or distributed among Bundibugyo and Taï forest Ebolaviruses with multiple occurrences (Figure 6D). AIMLASYTV was more specific to Reston and Sudan Ebolaviruses with multiple occurrences while the other epitopes were either specific to Sudan Ebolavirus or Marburg virus with unique occurrences (Figure 6E).

Frequency of antigenic peptides for both VGP and MXP were dominant for HLA*A02:01 and HLA*A02:02 alleles (A) with VGP frequency higher in the latter than in the former and vice versa. Epitope distribution for the different immune alleles showed that antigenic sites for each genomic structure (VGP and MXP) could be recognised by particular immune alleles while others were recognized by all (B, VGP and C, MXP). The identified epitopes were seen to occur variably among species with some epitopes being specific to a given viral specie and others common to a species (D, VGP and E, MXP).
Several epitopes were predicted for HLA class II-DRB alleles; VGP (n = 221) and MXP (n = 154) showing high frequencies for DRB1*01:01 and DRB1*01:04 alleles (Figure 7A, Table S3) with MXP higher for the latter than the former and vice versa for VGP. The four epitopes with highest VGP frequencies and coverage (COV) per HLA-DRB1 allele were; IILFQRTFSIPLGVI (7.05%) & COV = 6/8, RWGFRSGVPPKVVNY (3.52%) & COV = 3/8, WVIILFQRTFSIPLG (3.52%) & COV = 3/8 and IILFQRAISMPLGIV (3.40%) & COV = 6/8 (Figure 7B). The four epitopes with the highest MXP frequencies and coverage per HLA-DRB1 allele were; PQYFTFDLTALKLIT (7.25%) & COV = 5/8, ITHFGKATNPLVRVN (4.38%) & COV = 5/8, TTAAIMLASYTITHF (3.00%) & COV = 2/8 and QKTYSFDSTTAAIML (2.75%) & COV = 2/8 (Figure 7C). Based on VGP and HLA-DRB1 allele epitope distribution per Ebola species; IILFQRTFSIPLGVI was dominantly Zaire Ebolavirus, WVIILFQRTFSIPLG and RWGFRSGVPPKVVNY were solely Zaire Ebolavirus and IILFQRAISMPLGIV was dominantly Reston Ebolavirus (Figure 7D). For the MXP epitopes and HLA_DRB1 allele distribution per Ebola species; PQYFTFDLTALKLIT was relatively equally distributed among the different Ebolaviruses, ITHFGKATNPLVRVN was solely Zaire Ebolavirus, TTAAIMLASYTITHF and QKTYSFDSTTAAIML were dominantly Zaire Ebolavirus (Figure 7E).
Frequency of antigenic peptides for both VGP and MXP were dominant for DRB1*01:01 and DRB1*01:04 alleles (A) with VGP frequency higher in the former than in the latter and vice versa. For the top four selected Epitopes, frequency distribution for the different immune alleles showed that antigenic sites for each genomic structure (VGP and MXP) could be recognised by particular immune alleles, others were recognized by all and some could not be recognized by any of the immune alleles (B, VGP and C, MXP). The identified epitopes were seen to occur variably among species with some epitopes being specific to a given viral specie and several others common to a species and for some species, none had the epitope site (D, VGP and E, MXP).
Several epitopes were predicted for HLA class II-DQA-B alleles; VGP (n = 41) and MXP (n = 11) showing high frequencies for HLA-DQA1_03_01-DQB1_03_01 and HLA-DQA1_05_04-DQB1_05_03 (Figure 8A, Table S4) with VGP higher for the former than for the latter. The four epitopes with highest VGP frequencies per HLA-DQA-B allele coverage were; ETEYLFEVDNLTYVQ (13.55%) & COV = 3/9, FHKEGAFFLYDRLAS (8.64%) & COV = 1/9, RGVNFAEGVIAFLIL (7.24%) & COV = 3/9 and WVIILFQRTFSIPLG (6.78%) and COV = 1/9 (Figure 8B). The four epitopes with highest MXP frequencies per HLA-DQA-B allele coverage were; YSFDSTTAAIMLASY (33.33%) & COV = 2/7, HTPSGVASAFILEAT (33.33%) & COV = 5/7, IYSFDSTTAAIMLAS (8.33%) & COV = 1/7 and HTPSGVASAFILEAK (8.33%) & COV = 4/7 (Figure 8C). Based on VGP epitope and HLA-DQA-B distribution per Ebola species; ETEYLFEVDNLTYVQ and FHKEGAFFLYDRLAS was dominantly Zaire Ebolavirus, RGVNFAEGVIAFLIL was dominantly Sudan and Reston Ebolaviruses and WVIILFQRTFSIPLG was solely Zaire Ebolavirus (Figure 8D). Based on MXP epitope and HLA-DQA-B distribution per Ebola species; YSFDSTTAAIMLASY was relatively distributed equally among the different Ebolaviruses, HTPSGVASAFILEAT was dominantly Reston Ebolavirus, IYSFDSTTAAIMLAS was solely Reston Ebolavirus and HTPSGVASAFILEAK was equally distributed between Reston and Taï Ebolaviruses (Figure 8E).
Frequency of antigenic peptides for both VGP and MXP were dominant for HLA-DQA1_05_04-DQB1_05_03 allele with that of VGP higher than MXP but only MXP presented a high frequency for HLA-DQA1_03_01-DQB1_03_01 allele (A). For the top four selected epitopes, frequency distribution for the different immune alleles showed that antigenic sites for each genomic structure (VGP and MXP) could be recognised by particular immune alleles, others were recognized by all and some could not be recognized by any of the immune alleles (B, VGP and C, MXP). The identified epitopes were seen to occur variably among species with some epitopes being specific to a given viral specie and several others common to a species and for some species, none had the epitope site (D, VGP and E, MXP).
Based on the data collected, peptide scores and peptide frequencies expressed in the viral population (simulating host population) were used to calculate the transmission intensity ratio predicting how fast or slow the virus will spread within its host population (Table 2). The majority of the MHC HLA class I peptides showed a value greater than 1 while the majority of the MHC HLA class II peptides showed a value less than one. In general, TRI < 1, showed multiple expressed epitopes within each species and high distribution within the population and vice versa for TRI > 1. Zaire and Reston Ebolaviruses showed the lowest TRIs in the selected data.
Epitopes predicted for MHC class I and II were predicted and their antigenic scores (PS)/peptide frequency within the host population were analyzed to determine values which could be used to determine epidemic outcomes of the disease. PS/PF > 1 was considered high transmission risk value. M = mixed, S = specific.
In this study, Ebolavirus amino acid sequence data was obtained for Zaire, Sudan, Côte d'Ivoire (Taï forest), Uganda (Bundibugyo) and Philippines (Reston) species. These sequences were subjected to phylogenetic analysis, which took into consideration all known functional motifs and domains for each sequence to construct the tree. It was observed that, species variation was higher for VGP compared to Nuclear and Matrix proteins of the viral genome. This observation could have resulted from pairwise alignments, whose identity scores accounted for species specific clustering observed.
The first two Ebolavirus species, Zaire and Sudan, identified in 1976 and later, Reston and Côte d'Ivoire, in 1989 and 1994, respectively, revealed after comparison that the four viral genomes differed from each other by 36.7–42.3% (Towner et al., 2008). In 2008, Uganda Ebolavirus species was identified and its genomic sequence differed from previously known viruses by 31.7–42.4% (Towner et al., 2008). Work done by Leroy et al. (2002) showed that the mean amino acid distances between the same NP subtypes was about 45%, compared to 65–66% in VGP. These values were higher than those observed in this work with probable difference being that we performed an amino acid sequence alignment, whilst the authors carried out a nucleotide sequence alignment. The longest branch on the tree associated with Marburg virus suggest an ancestral lineage which could be a consequence of evolutionary time wherein, Marburg virus and Ebolavirus were isolated in 1967 (Siegert et al., 1967) and 1976 (Bowen et al., 1977; Johnson et al., 1977), respectively. They also show a genomic difference ≥50% at the nucleotide level (Towner et al., 2006). The presence of one gene overlap in Marburg virus compared to several in Ebolavirus delineates the main genomic difference between them (Kuhn, 2008).
The observed functional relationship between related hosts (mammal) and Ebolavirus proteins, identified within the interactome, showed direct interactions between the former, but none was observed between VP40 and ENV (gp160), and likewise to host proteins. Given the identification of gag, pol, and env (ENV) retroviral genes, the pol gene, which encodes reverse transcriptase (RT), is considered the most conserved of the retroid elements (Simmons et al., 2002). The polypeptide encoded by the env gene is cleaved into two proteins; a surface protein for receptor recognition, and a transmembrane subunit, which anchors the ENV complex to the membrane. The ENV transmembrane unit interacts directly with the cell membrane, favoring viral penetration (Bénit et al., 2001) into the cell which has been elucidated for human immunodeficiency virus type 1 (HIV-1) (Connolly et al., 1999; Xu et al., 1998) and Ebolavirus (Sanchez et al., 2001). The ENV-dependent pathways for HIV accounts for almost 50% of antigen presentation mediated principally by the lectin DC-SIGN (CD209). CD209 molecules, form part of dendritic cells equipped with molecular diversity for HIV-1 capture, which binds the viral envelope gp120 with high affinity. Efficient cross presentation of HIV antigens from incoming virions activates anti-HIV CD8+ cytotoxic T lymphocytes. The major role of CD209 in viral entry into the host cell, solidifies its identity within the predicted interactome. Though the observed functional roles include intracellular virion transport, protein signaling and peptide identification, associated with CD209, no direct interaction was observed with the viral structural genes predicted on the network. This could suggest a domain specific function in CD209 for viral-host interactions, and though similar in Ebolaviruses, the functional pathway may be different.
The Ebolavirus VGP is synthesized as a secreted (sGP) or full-length transmembrane form, having distinct biochemical and biological properties. Multiple lines of evidence suggest that VGP plays a prominent role in clinical manifestations of Ebolavirus infection. A change of immune response could result from inhibition of neutrophil activation by sGP, while the transmembrane form could contribute to symptoms of hemorrhagic fever due to viral attack of reticuloendothelial and blood vessel cells (Ströher et al., 2001; Yang et al., 1998). The different host cell targets and roles played by sGP and the transmembrane form, makes them potential targets that could be exploited for control of Ebolaviral burden. The type of preferred cells by each VGP form, should determine the type of pathogenic manifestation and nature of transmission intensity observed within the population. Therefore, drug targets common across viral species or specific to most virulent forms will be vital for quick clearance of an infection.
The assembly and budding of Ebolaviruses from the cell membrane is a process directed by the viral matrix protein, VP40 (Harty et al., 2000; Panchal et al., 2003). VP40 alone is capable of assembling and budding filamentous virus-like particles from cells (Geisbert & Jahrling, 1995; Johnson et al., 2006; Noda et al., 2002). The specific interaction of VP40 with membrane-associated VGP and VP24 during the budding process, is a possible occurrence. Work done by Bornholdt et al. (2013), showed that binding of VP40 to RNA forms a ring structure at a perinuclear location. Observation of these rings was only in infected cells, but not mature purified Ebolaviruses (Gomis-Rüth et al., 2003), which suggests that, the ring structure was not necessary for matrix assembly. Therefore the VP40 RNA-binding rings could possess a critical function within infected cells (Gomis-Rüth et al., 2003; Hoenen et al., 2010). Previous data showed that, recombinant viruses that have an RNA-binding knockout mutation for VP40 (R134A) failed to replicate (Hoenen et al., 2005), suggesting a dual role for VP40, which includes: viral transcription with assembly, and budding, for the release of new viral particles. Given that VP40 is structural and shows possible interactions with VGP, suggests a contributory role to host immune activation pathway, and though directing different cellular processes, their immune-interaction makes them potential targets for vaccine design and control of Ebolavirus disease.
The close relationship between Ebola genomic structures; VP40, CD209 and ENV and between HIV-1 and Ebolavirus, motivated our use of VP40 and VGP for the prediction of antigenic peptide epitopes required for generating potent host antibodies against the virus. The interactive nature between ENV and CD209 proteins, could explain the downstream signaling reactions necessary for cytokine activation. Identifying target signaling molecules activated by interaction of CD209 with Ebolavirus antigens, could suggest a possible path for curbing the loss of body fluids during epidemic outbreaks. The presence of LCK and its kinase function suggests a protein signaling role in driving the infection and loss of fluid pathway associated with Ebolavirus. Its involvement in CD4 and CD8 binding makes its pathway, an interesting target area for Ebolavirus disease control. The signaling of T cell receptors (TCR), which results from peptide-MHC binding, requires the phosphorylation of immunoreceptor, tyrosine-based activation motifs (ITAMs) and CD3 complex, associated with SFK Lck (Palacios & Weiss, 2004). Generally, it is accepted that, the activation of T cells favors Lck activation by pMHC binding. Given the stable association of Lck to CD4 and CD8 coreceptors (Rudd et al., 1988; Veillette et al., 1988), other interaction models propose that MHC binding activates Lck by trans-autophosphorylation. Basing on this information, the role-played by LCK within the network and its interaction with immune cells becomes very clear. The implication of the LCK gene in Ebolavirus disease intensity could be related to a hyper activation cascade leading to massive release of cytokines (Cytokine storm), which is an event common to patients with septic shock (Mackenzie & Lever, 2007). The Lck protein kinase pathway identified in the network as a cluster, showed interaction with proteins including SRC (Proto-oncogene tyrosine-protein kinase), MERTK (Tyrosine-protein kinase), TYRO3 (Tyrosine-protein kinase receptor), UFO (Tyrosine-protein kinase receptor), ACK1 (Activated CDC42 kinase 1), BMPR2 (Bone morphogenic protein receptor type-2), YES (Tyrosine-protein kinase) and FYN (tyrosine protein kinase). The UniProt Knowledge Base (KB) (www.uniprot.org), associates these proteins to downstream ligand binders and signal transducers, which activates other proteins in a phosphokinase related interaction type. Several of these are involved in disease pathways including SRC (colon carcinoma), MERTK (retinitis pigmentosa, MIM: 613862), UFO (thyroid tumorigenesis) and BMPR2 (pulmonary hypertension primary, PPHL 1, MIM: 178600). This cluster suggests that, the role played by LCK in kinase function and implicates it to epidemic levels observed for Ebolavirus disease.
MiRNAs are potentially known to regulate several proteins (Grimson et al., 2007) and also modulate their concentrations in a dose-dependent manner (Baek et al., 2008; Selbach et al., 2008). After Grigoryev et al. (2011) identified several miRNAs significantly differentially expressed in T-lymphocytes, they showed a decrease in mRNA expression for target genes of upregulated miRNA molecules. This was confirmed by inhibiting two of these miRNA targets (miR-221 and miR-155) in CD4+ cells, which increased lymphocyte proliferation by liberating four (PIK3R1, FOS, IRS2, and IKBKE) gene targets, hence favouring their proliferation and survival. MiR-155 overexpression has been shown to increase in vivo antiviral CD8+ T cell responses (Gracias et al., 2013). Among the highlighted miRNA gene targets above, IKKE (IKBKE) was the only one captured in this study and identified in the top three proteins. MiRNAs and their related disease pathway, hsa05131 (microbial infection, shigellosis) and cellular process, hsa01430 (cell communication), could suggest the possibility of successful viral transmission and disease occurrence through gene regulation within each pathway. Knowledge of other regulatory elements such as transcription factors and drug targets for related genes within the network, suggests a complex regulatory platform for the control of Ebolavirus disease. Transcription factors to gene targets like CD209 known for antigen binding and immune presentation could be further molecularly analysed as potential targets for gene regulation and disease prevention within the host.
Gene products in the HLA region (chromosome 6) facilitate antigenic protein processing and peptides presentation to CD8+ cytotoxic T lymphocytes (CTLs) and CD4+ T helper cells (Coffin, 1996; Dyer et al., 1997). HLA allele’s multiplicity has favoured great species diversity as individuals differentially adapt to the wide range of microbial agents such as HIV-1. Optimal immune defence against HIV-1 could depend on the combined intervention of CD4+ and CD8+ lymphocytes (Rosenberg et al., 1997) wherein each polymorphic class I and class II loci contributes separately to the resultant immune response. CD209 favors specificity of HIV antigen presentation to MHC-II alleles while dendritic cells promote HIV-specific CD4+ cells activation and eventual infection. Similarity between HIV-1 and Ebolavirus in the use of antigen-immune interactions to penetrate the host cell could be seen from related genes identified in the network. Components of the immune system with protective role against infection of Ebolavirus are not clearly known due to the fact that the pathogen replicates at unexpected high rates, overwhelming host infected cells and immune defenses with related protein synthesis mechanisms (Sanchez et al., 2001).
From our study, HLA class I peptide antigenicity and frequency values for VGP and MXP within the pathogen population, showed no significant statistical difference. Their specificity was tied to HLA*A02:01 and HLA*A02:02 alleles for all the validated peptides, unlike differential selectivity observed for other tested host alleles. This was in conformity with host expression of HLA*A02 allele against the HIV-1 virus expressed in the Botswana (Novitsky et al., 2001) and Rwandan population (Tang et al., 1999). The FLLQLNETI epitope for VGP was specific to the Zaire Ebolavirus with a single occurrence while the other VGP and all the MXP epitopes were related to other viral species with multiple occurrences. Although the Zaire strain has been considered the most lethal (Feldmann et al., 1994; Sanchez et al., 1996), the observed transmission intensity (TRI) for the FLLQLNETI epitope was relatively low compared to the FLTNTIRGV epitope for VGP (Bundibugyo and Taï forest), with a TRI twice that of the former. This brings two questions to mind; 1) Is the Ebolavirus epidemic caused mainly by the Zaire Ebola species? and, 2) What causes the sudden change in antigenicity that results in epidemic trends currently observed in West Africa (Liberia, Sierra Leone and Equatorial Guinea). One possible solution could be due to the high phylogenic interspecies differences observed within the VGP genomic structure, suggesting a species specific (pairwise identity) pathogenic driven occurrence. The grouping of Zaire Ebolavirus species with other related species and not vice versa could suggest a derivation of other species from the Zaire virus, hence the host immunity may have adapted to keeping the Zaire strain below epidemic levels. Introduction of high antigenic variants from related strains could be the driving force of the epidemic levels observed.
HLA class II antigenic against frequency values for identified epitopes, presented with high statistical differences for VGP and MXP genomic structures. The validated peptides showed high antigenic values for HLA II thus favouring a high TRI. The most common host allelic frequencies for VGP and MXP were HLA-DRB1*01:01 and HLA-DRB1*01:04 and were consistent with that of the Botswana population carrying the HIV-1 virus though different for other alleles observed with high frequencies in that population. The viral species identified for selected HLA-II peptide epitopes were predominantly and specifically Zaire Ebolavirus. All epitopes presented with low TRIs except for QKMYSFDSTTAAIML which had a TRI > 10 for the MXP genomic structure. This was contrary to the HLA-I high TRI data observed for VGP genomic structure of the Zaire Ebolavirus. Given that specificity of antigen presentation to HLA-II immune proteins is facilitated by CD209, MXP should contribute highly to immune response and signal transmissions downstream. MXP has not always been associated with antigenic interactions but the fact that it could interact with VGP at the viral surface makes its contribution to immune response of considerable importance.
Antigenicity of peptide epitopes for HLA-DQA-B were statistically significant for VGP and not MXP. The VGP validated epitopes were dominant for Zaire Ebolavirus species while those of MXP were dominant for Reston Ebolavirus. Both genomic structures showed high frequencies for HLA-DQA1_05_04-DQB1_05_03 while only frequencies for MXP epitopes were high for HLA-DQA1_03_01-DQB1_03_01. Only HLA-DQA1-B1*03 was in conformity with the alleles identified for the Botswana HIV-1 infected individuals. This allele was specific to MXP and dominantly Reston Ebolavirus, suggesting a possible contribution to immune activation. The study from Botswana genotyped humans for HLA-II allele frequencies and from our study suggests immune specificity to different Ebola genome structures. This observation highlights the difference in antigenicity between the Ebolavirus and HIV-1 virus. Though the low TRI and lack of significant difference for MXP epitopes, the observed antigenic specificity with that of VGP genomic structure could play vital roles for viral pathogenesis within the host.
The multiple occurrences of several antigenic epitopes for a given viral species could suggest repeat sequences in tandem within the genome known as retrotransposons, which favor high genome variation and transmission. Previous work done by several authors on the similarity of reverse transcriptase (pol gene) in retroelements including retrotransposons and retroviruses such as Ebolavirus, suggest that retroviruses could have evolved from long terminal repeat (LRT) retrotransposons by acquiring a functional env gene (McClure, 1991; Xiong & Eickbush, 1990). Therefore, the env gene (VGP) should be critical for viral transmission in its host and could be targeted with anti-retroviral technology currently used in HIV patients. Its identification in the interactome suggests an interaction with several human proteins involved in signal transduction. The interplay between the MXP, VGP and their related interacting human proteins makes all these various genes potential targets for Ebolavirus control through drug/vaccine design or the use of genetic technology through interference of viral replication.
This study is the first of its kind that has exploited all possible viral antigenic targets and possible systemic routes in the host that could be targeted individually or together for the control of Ebolavirus disease. From this study, the phylogenetic relationship distinguished in the viral envelope of Ebolaviruses and their similarity to HIV-1 viral genomic structures shows that common pathways to pathogenic manifestations within the host are eminent. The ability to clearly understand cellular pathways driving pathogenicity is critical for arresting rapid and abundant cytokine activation leading to shock and organ malfunction. Though the Zaire Ebolavirus has been identified as the most virulent; the TRI for this species generally shows a low transmission rate compared to others, which is in accordance with viral-host cohabitation and adaptation. This suggests that, epidemic occurrences of the Ebolavirus disease could result from related species which have no genetic adaptation with its host. The sequence repeats predicted within antigenic epitopes, occurred more for the non-Zaire Ebola viral species. The identified antigenic epitope targets for this species could be used for drug/vaccine designs. Potential microRNA’s, transcription factors and known drugs identified for most gene targets, suggest possible points in the viral pathogenic pathway that could be the focus of modern genetic technology for arresting viral replication and epidemics.
The Geneious software (V.6.1.8) (Kearse et al., 2012) and its routine plugin packages were used for extracting and analyzing obtained sequences for various Ebolavirus species. The key word “Ebolavirus”, was used to search the UniProtKB (Magrane & Consortium, 2011) (Table S1), resulting in 348 protein sequence variants (database searched in September, 2014), from the five main regional strains highly associated with Ebola Hemorrhagic Fever (Dowell et al., 1999), which are: Zaire Ebolavirus, Sudan Ebolavirus, Bundibugyo Ebolavirus (from Uganda), Taï Ebolavirus (from Coté d’Ivoire) and Reston Ebolavirus (from the Philippines). The sequences also included related mammalian protein sequences (Homo sapiens, Mus musculus, Rattus norvergicus) associated with the viral genome based on genetic motifs, domains and Gene Ontology (GO) terms (Ashburner et al., 2000) captured in various phyla. Sequences were aligned using a novel method for rapid multiple sequence alignment based on fast Fourier transform (MAFFT V7.017) (Katoh et al., 2002). The parameters involved an auto alignment algorithm, a Blosum 62 (Mihalek et al., 2007) scoring matrix, a gap penalty of 1.5 and an offset value = 0.123. The aligned sequences were then subjected to a Geneious tree builder which uses pairwise genetic distances from the sequence alignment, for tree construction, while basing on the following parameters: Jukes & Cantor (1969) genetic distance model for amino acid substitution, Unweighted Pair Group Method with Arithmetic mean (UPGMA) (Yu-Shiang et al., 2014), for tree building and jacknifing (Jones, 1974) while sampling for the best tree (random seed = 932, 120; replicates = 500; support threshold = 80%). Sequence clusters identified from the tree were exported in FASTA format for MHC HLA class I and II epitope predictions. UniProt accession numbers were extracted and used for protein interaction predictions.
UniProt accession numbers for the 348 sequences were loaded into Cytoscape software (V.2.8.1) (Shannon et al., 2003), a bioinformatics platform for molecular interaction networks visualization with integration of identified interactions to other biological data, and its routine plugins were used to predict and analyze the network. APID2NET (Hernandez-Toro et al., 2007), was used to predict the protein interaction map. This tool queries interactome data from the Agile Protein Interaction Data Analyzer (APID) server (http://bioinfow.dep.usal.es/apid/) providing an interactive analysis of individual protein-protein interaction information into a network. During the interaction, the program considers parameters like GO terms, InterPro domains (Hunter et al., 2012), PFAM domains (Bateman et al., 2002), experimental data for validated interactions and all necessary genetic information captured in the APID database for each protein. Network Analyzer, a Cytoscape plugin was used to evaluate node attributes for the complex network and it considers parameters such as: degree distribution (Barabási & Oltvai, 2004; Diestel, 2005), neighborhood connectivity (Maslov & Sneppen, 2002), shortest paths (Watts & Strogatz, 1998), clustering coefficients (Barabási & Oltvai, 2004), shared neighbors, topological coefficients (Stelzl et al., 2005), stress centrality (Shimbel, 1953), betweenness centrality (Brandes, 2001) and closeness centrality (Newman, 2003) to name a few. The Biological Network Genes Ontology tool (BiNGO) was used to determine full GO terms significantly overrepresented in our gene set (Maere et al., 2005) with species target Homo sapiens. The plugin CyTargetLinker (Kutmon et al., 2013) was used to extend the interaction network with regulatory elements such as: miRNA-target (Hsu et al., 2014), drug-target or TF-target interactions and TargetScan (Friedman et al., 2009) to identify genes regulated by these elements. Identified microRNA molecules were associated to particular pathways based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2014).
The various Ebolavirus sequence sub clusters, [virion envelope glycoprotein, VGP (N = 63) and Matrix protein, MXP (N = 19)] were separately submitted for: 1) MHC class I epitope predictions at the HLA restrictor 1.2 Server (Erup Larsen et al., 2011), a tool for patient-specific predictions of HLA restriction elements and optimal epitopes (8 – 11 amino acids) within peptides and 2) MHC class II epitope predictions at Net MHCIIpan 3.0 server (Karosiene et al., 2013) which predicts binding of peptides (9 – 19 amino acids long) to MHCII alleles for three human isotypes (HLA-DR, HLA-DP and HLA-DQ). Given the vacuum of information on the MHC HLA class I & II allele types in Ebola stricken populations, selected alleles associated with the HIV virus in African related populations (Tang et al., 1999; Novitsky et al., 2001) were considered for use in this study since both viral species have been shown to possess the homologous hairpin structural envelope glycoprotein (gp160) (Watanabe et al., 2000), which is a target for several HIV vaccines. Based on previous work done by our group (Ojwang et al., 2014), nine monomers predictions were better preferred for MHC class I, HLA-A alleles while 15 monomers were better preferred for MHC class II, HLA-DRB and HLA-DQ alleles (Table 3).
Nine monomers and fifteen monomers were predicted for HLA-I and II alleles respectively.
Peptides were selected on two criteria; threshold affinity for strong binders (0.5 for percentage rank and 50 for IC50) and threshold affinity for weak binders (1 for percentage rank and 100 for IC50). Only strong binders were considered for further analysis. Files containing strong binder epitopes for each Ebola structural genomic class variant and for each species was generated in FASTA format and submitted to Vaxijen software (V.2.0) (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). Viral antigenic prediction filter was used at a cutoff of 0.5 and all epitopes with antigenicity values (peptide score) ≥ 0.5 separately for MHC class I and II alleles were further considered for analysis.
For every validated epitope in our data, the peptide scores and peptide frequencies were considered for the calculation of a novel value called TRANSMISSION INTENSITY (TRI) which translates how high or low the disease transmission will develop into an epidemic situation within the population. The value was based on the ratio of peptide score (PS)/peptide frequency (PF). If TRI was around one, it means the transmission is stable and could easily be contained to elimination. If TRI > 1, it means, the transmission poses a bigger risk to the infected individuals given that high antigenicity will activate a high kinetic and signalling response pathway (Yamashita et al., 1998) reaching epidemic levels due to the host immune system generating high antibody levels against the virus possibly leading to hypotensive shock, characterized by; fever, myalgia, malaise, severe bleeding and coagulation abnormalities, including gastrointestinal bleeding, rash, and a range of hematological irregularities, such as lymphopenia and neutrophilia (Colebunders & Borchert, 2000). If TRI < 1, it means, the transmission poses a low risk within the population and could easily be eliminated by the host immune system. Given that antigenicity is low and peptide frequency is high, the host quickly develops immunity against the pathogen. The different transmission intensities will depend on the MHC HLA allele type and its frequency within the population.
Batch analysis for evaluating node attributes for the network was used to fit the best line between the data points using the least squares method (Weisstein, www.mathworld.wolfram.com). GO terms for the predicted interactome were validated based on the Hypergeometric test after Benjamini & Hochberg False Discovery Rate (FDR) (Benjamini & Hochberg, 1995) correction and at a significance level of 0.05. Files for each structural genomic class variant per species was used to evaluate frequency distribution for epitopes against peptide scores, HLA allele frequency and frequency within each species protein sequence (supporting Table S2, Table S3 and Table S4). After normalizing every value to the largest in each dataset (epitope peptide antigenic score and epitope peptide frequency) of different Ebola sequences, paired student one tailed t-test was performed to compare means between the groups (α = 0.05). This was to evaluate how epitope targets were distributed across different Ebola species and their epidemiological implications.
The complete list of UniProt KB protein sequence codes for extracted Ebolavirus sequences used for analysis can be found in Table S1.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)