Keywords
Fenofibrate, cancer, PPAR, angiogenesis, metastasis, prostate, cancer, glioblastoma, melanoma, nelfinavir
Fenofibrate, cancer, PPAR, angiogenesis, metastasis, prostate, cancer, glioblastoma, melanoma, nelfinavir
Version 2 contains a detailed description of fenofibrate activity on cell membranes and mitochondria. It also highlights differences between fenofibrate and fenofibric acid regarding pharmacological activity on cancer tissues. Figures 1 and 8 have been added for a better understanding of these issues. Table 6 was modified updating references. Reference 158 was also modified and 2005 was replaced by the correct year: 2015. Also, ten new references were added.
See the author's detailed response to the review by Krzysztof Reiss
See the author's detailed response to the review by Jarosław Czyz
FF: Fenofibrate /HUVEC: Human vascular endothelial cells /MCP-1: Monocyte chemotactic protein-1 /VCAM-1: Vascular cell adhesion molecule 1 /ICAM-1: Intercellular adhesion molecule-1 /VEGF: Vascular endothelial growth factor /VEGFR: Vascular endothelial growth factor receptor /HIF: Hypoxia inducible factor /PPAR: Peroxisome proliferator activator receptor FGF: Fibroblast growth factor /bFGF: basic fibroblast growth factor /TSP-1: Thrombospondin-1 /RXR: Retinoid X receptor /ATRA: All transretinoic acid /ER: Endoplasmic reticulum /RCC: Renal cell carcinoma /PDGF: Platelet derived growth factor /TRIB-3: Tribbles homolog 3 /IGF: Insulin like growth factor /PEG2: Prostaglandin E2 /CTMP: C-terminal modulator protein (which inhibits AKT phosphorylation) /SCC: Squamous cell carcinoma /SREBP: Steroid regulatory element binding proteins /FAS: Fatty acid synthase.
Fenofibrate (FF) is a drug of the fibrate class (a fibric acid derivative) (for chemical structure, see Figure 1) that has been used since 1975 to reduce cholesterol (LDL and VLDL) and triglyceride levels and increase HDL in patients at risk of cardiovascular disease and for treatment of atherosclerosis (1 and 47). FF is one of the most commonly prescribed fibrates, and has a well known efficacy and tolerability profile1.
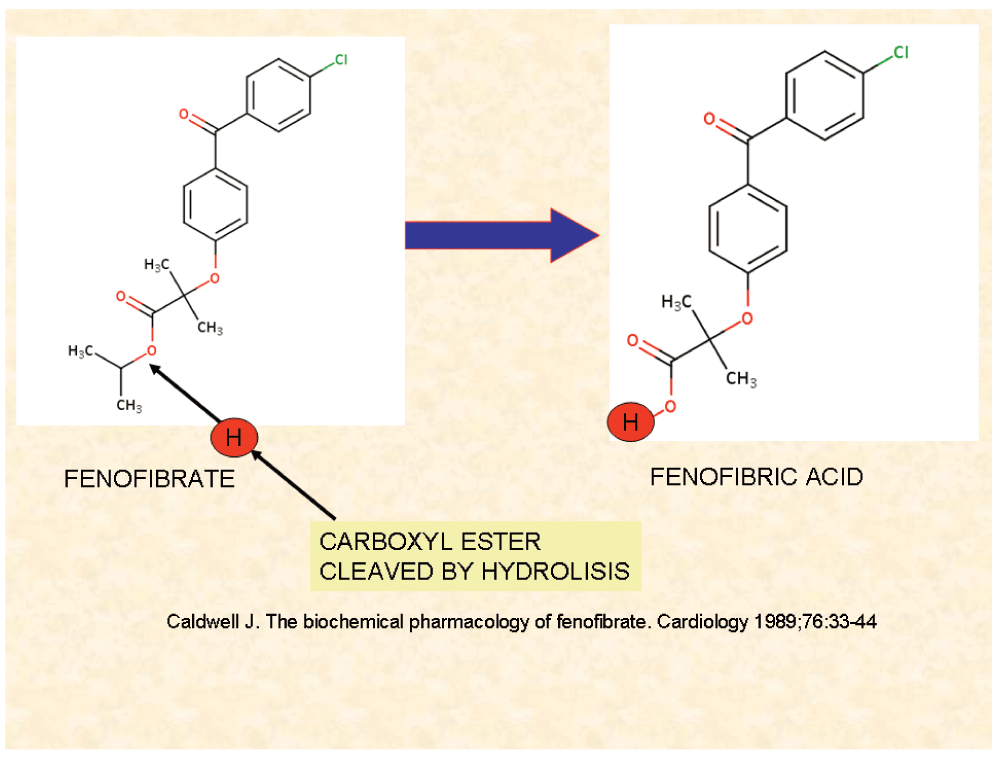
Unprocessed FF is capable of interacting with biological membranes. Fenofibric acid is a potent PPARα agonist that does not interact with membranes.
FF seems to lower lipid levels by activating peroxisome proliferator-activated receptor alpha (PPARα), a nuclear receptor which acts as a ligand activated transcriptional factor and activates lipoprotein lipase and reducing apolipoprotein CIII expression. These activities increase lipolysis and eliminate triglyceride-rich particles2.
PPARs are a widely distributed family of nuclear receptors. Three isoforms have been identified: PPARα, PPARβ/δ, and PPARγ. Ligand binding activates these receptors that play key roles in cellular energy homeostasis, modulating glucose and lipid metabolism. PPARα as illustrated in Figure 2, is the molecular target of the fibrate class of drugs, which act as agonistic ligands of PPARα (Figure 3). Other fibrates like clofibrate and bezafibrate are also ligands for this receptor. Poly-unsaturated fatty acids are the natural ligands.
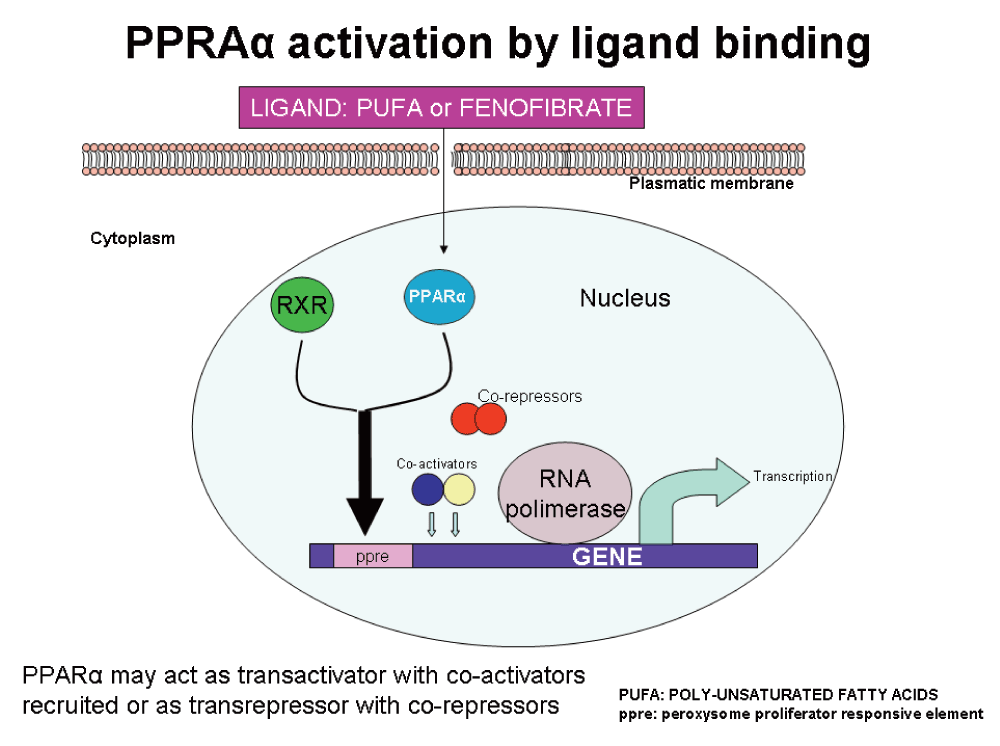
Upon ligand binding, PPARα dimerizes with RXR (retinoic X receptor) and both interact with peroxisome proliferators responsive elements of the target gene. Coactivator proteins and RNA polymerase are recruited and the transcription machinery is set to work (trans-activation). When co-repressor molecules are recruited, trans-repression is unleashed and no transcription is produced.
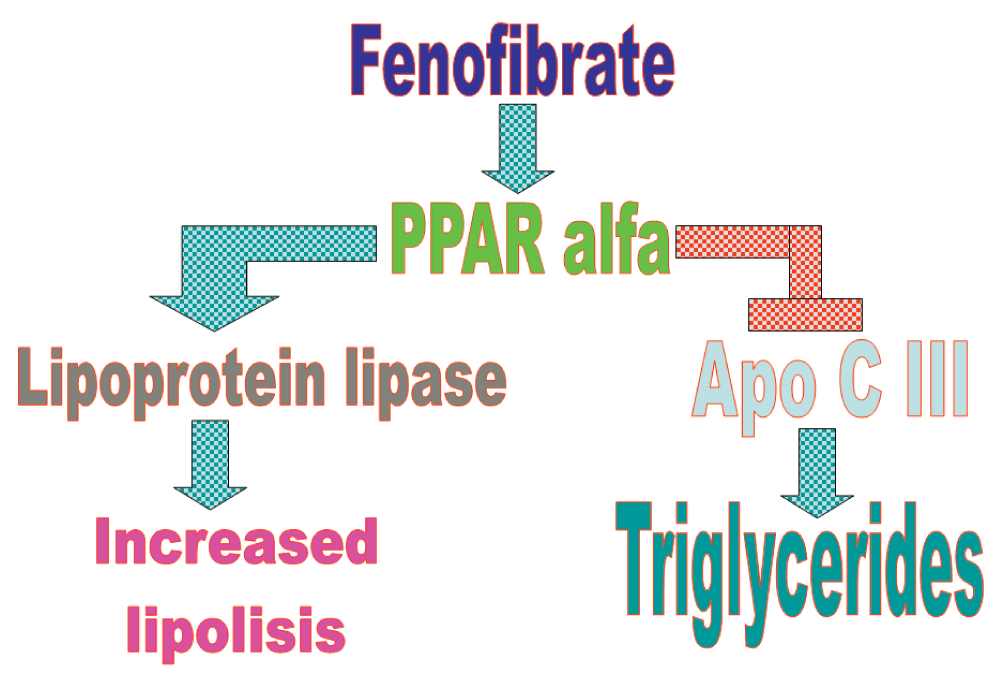
We shall not go any deeper into lipid metabolism activities of FF because our goal is to consider the effects of this pharmaceutical in cancer prevention and treatment rather than in cardiovascular risk.
Before the year 2000, all publications on FF considered anti-lipogenic properties of this drug with no mention of possible anti-cancer activity.
We only found two publications of FF activities before than that may be related with cancer:
In 2002 two findings on FF anti-cancer therapy were important:
FF has the capacity to induce hepatocarcinoma in rodents, but this effect seems specific for this species, as in humans it has been shown to have cytotoxic effect on HepG2 hepatoma cell line at high concentrations and in a dose dependent manner7.
Varet et al. (2003)8 demonstrated that FF inhibits angiogenesis in vitro and in vivo.
MCP-1 (monocyte chemotactic protein-1) is a protein that recruits and activates monocytes during inflammatory processes but also plays a role in cancer: it increases proliferation and invasion of CaP cells (prostate cancer)9. FF inhibits expression of MCP-1 on activated endothelial cells10.
PPARα agonists like FF were found to inhibit endothelial VEGFR211 expression.
In diabetic II hyperlipidemic patients, FF decreased E-selectin by 10% and ICAM-1 by 4% and no change of VCAM-1 was detected12.
But the first real hint towards a possible anticancer activity of FF was provided by Holland et al. in 200413 who showed by transcriptome analysis of endometrial cancer cells an overexpression of PPARα. This finding led them to the investigation of FF activity on tumor cells. FF reduced proliferation and increased apoptosis of cancer cells. At the same time, a second publication14 showed the FF potential to reduce metastasis of melanoma cells in an experimental setting.
In ooforectomized rats, treated with estradiol and FF for 30 days, the uterine mass decreased, uterine glands had normal structure and there were no cases of atypical hyperplasia15.
Kubota et al.16 found that apoptosis induced by FF in cultured human hepatocytes was due to caspase-dependent apoptosis by inhibiting phosphorylation of Akt, in a PPARα independent manner.
The role of chemokines produced by different stromal cells stimulating proliferation and angiogenesis in cancer tissues is well known. FF exerts a monocyte suppressing activity and reduces secretion of IL-6 and MCP-117.
Studying the possible anti-rheumatic activity of FF it was observed that this compound inhibits NF-kB18.
After these preliminary hints indicating the FF possible anti-cancer activities of FF, great amount of research and publications were dedicated to this issue. We summarized these findings in table 1 to table 15 according to anti-cancer activity disclosed.
On artificial grounds, but for better understanding, we have presented the anti-cancer activity of FF according to the main pro-tumor factor/pathway affected by the drug.
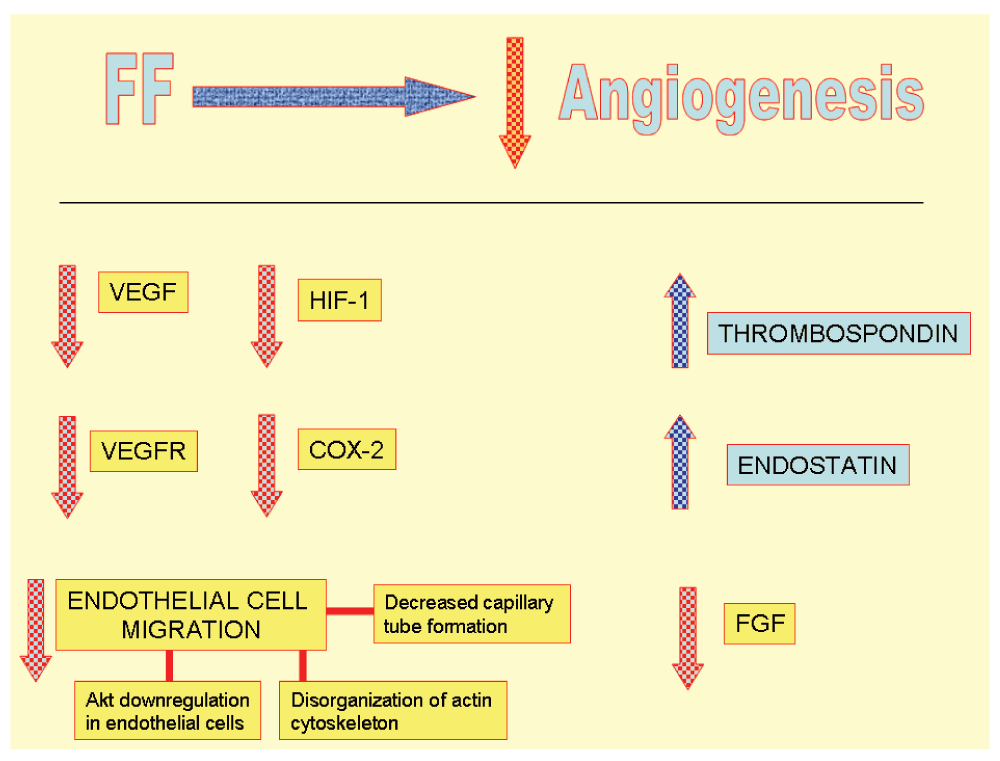
In the field of angiogenesis there is clinical experience with FF besides the laboratory experimental setting. In the research by Blann et al.19, hyperlipidemic patients treated with FF showed reduced lipidemia and plasmatic VEGF. No changes in VEGFR levels were seen. (Antiangiogenic mechanisms are summarized in Figure 4).
There is a tissue where FF has a pro-angiogenic effect: human retina. An anti-apoptotic property of FF in human retinal endothelial cells was reported by Kim J et al. in 200724. FF potently activated AMP-activated protein kinase (AMPK) and vascular endothelial growth factor (VEGF) mRNA expression. This finding was corroborated by the ACCORD medical study25 in patients with diabetes type 2 where FF was shown to have protective activity in diabetic retinopaty and other diabetic microvascular complications, probably through a decrease of human retinal endothelial cells apoptosis26.
This may mean that FF has a tissue specific activity that needs further investigations.
Probably the strongest and clearest evidence of the FF anti-angiogenic activity comes from the research of Panigraphy et al.21 of the Judah Folkman team and the research by Dana et al.23 on HUVEC.
Angiogenesis inhibition as described in Table 1 is probably one of the main anti-tumor activities of FF. Nickkho-Amiry et al.27 showed that treatment of human endometrial cells with a PPARα agonist leads to reduced secretion of VEGF in addition to reduced proliferation. This was potentiated by RxR (Retinoid X receptor) agonist like ATRA and inhibited by a PPARα antagonist.
| Reference | FF activities |
|---|---|
| Blann AD, 200119 | The levels of plasma vascular endothelial growth factor and its receptor Flt-1 in patients with hyperlipidemia and atherosclerosis are lowered by fluvastatin or FF by down-regulation of VEGF and VEGFR. |
| Goetze S, 20026 | Both PPARα- and PPARγ-activators inhibited VEGF-induced migration of HUVEC. VEGF-induced Akt phosphorylation was significantly inhibited by both PPARα- and γ-activators. |
| Varet J, 20038 | FF inhibits angiogenesis in vitro and in vivo, decreases bFGF induced Akt activation and COX2 expression and inhibits endothelial cell migration (the latter is attributed to disorganization of the actin skeleton). |
| Pozzi A, 201020 | PPARα agonists down-regulate angiogenesis by down-regulating epoxyeicosatrienoic acids synthesis. |
| Panigraphy D, 200821 | FF produces many anti-angiogenesis effects and suppresses tumor growth through direct and indirect angiogenesis inhibition: inhibits endothelial cell proliferation, decreases VEGF, increases TSP-1 and endostatin. |
| Zhou J, 201222 | Activation of PPARα suppresses hypoxia-inducible factor-1α (HIF-1α) signaling in cancer cells. PPARα activation promotes HIF-1α degradation in these cells. This was further confirmed using proteasome inhibitors, which reversed PPARα-mediated suppression of HIF-1α expression under hypoxia. |
| Dana N, 201323 | In HUVEC a Matrigel essay showed that FF effectively inhibited angiogenesis. |
| Meissner M, 200411 | FF and other PPARα agonists inhibit VEGFR2 expression in HUVEC. |
In a rat model Onalan et al. showed that FF caused regression of new endometriotic implants due to decreased angiogenesis28.
FF was included in many multi-agent anti-angiogenic regimens. One consisted of FF, celecoxib, thalidomide with metronomic low dose cyclophosphamide and etoposide. Patients were less than 21 year old with recurrent or progressive tumors. Half of the patients obtained benefits (CR + PR + SD)29.
Other metronomic anti-angiogenic multidrug protocols included FF as one of the pharmaceuticals, particularly for children with embryonal brain tumors and other malignancies30–32.
The COMBAT Protocol33 included low-dose daily temozolomide, etoposide, celecoxib, vitamin D, FF and retinoic acid and was used in 74 children with advanced refractory/relapsed solid tumors with two years overall survival of 43%.
The use of FF as part of anti-angiogenic multidrug protocols especially in pediatric cancer is constantly increasing.
Using a PPARα agonist like Wy-14643 in mice injected with tumor cells showed that treated animals had a marked reduction in tumor size and vascularization34.
In summary: FF increases thrombospondin synthesis, endostatin generation, decreases VEGF, COX2 and VEGFR2 expressions and prevents endothelial cells migration21,35.
Apoptosis induced by FF is caspase-dependent. In the case of clofibrate, apoptosis occurs through caspase 2 and 3 activation and ER stress in Jurkat cells43. Similar results were observed in Yoshida AH130 hepatoma cells44.
PPARα is increased in high grade renal cell carcinoma (RCC), but this does not provide any information about the functional status of this receptor, because in RCC the inhibition of PPARα induces apoptosis and agonists produce little or no effect45.
In 1983 Pascal et al.46 investigated the cardiovascular and anti-arteriosclerotic activities of FF and demonstrated that FF inhibited platelet derived growth factor (PDGF) stimulating activity on growth of cultured smooth muscle. Ten years later Munro et al.47 showed that FF is not a specific inhibitor of PDGF because smooth muscle cells growth was equally growth-inhibited by FF when the culture was stimulated with fetal calf serum, PDGF or basic fibroblast growth factor (bFGF). Our conclusion based on these two publications is that FF is a growth inhibitor in general (as least regarding vascular smooth muscle).
Anti-proliferation activity of FF has been found in many non-tumor tissues besides vascular smooth muscle, e.g. mesangial cells48 through inhibition of PI3K/AKT and ERK1/2 signaling pathways or by overexpression of TRIB3 (tribbles homolog 3) which inhibits Akt phosphorylation and slows cell cycle or causes arrest in G1/S49. In lymphocytes, FF also up-regulates TRIB3 causing cell cycle arrest50,51.
Endothelin-1 is a protein that increases cardiac fibroblast proliferation. PPARα agonists inhibit cardiac fibroblast proliferation down-regulating endothelin-152. FF also reduced c-jun expression in cardiac fibroblasts53. Endothelin-1 is an activator of the p38 mitogen activated kinase cascade. FF down-regulation of endothelin-1 also down-regulates the MAPK cascade in cardiomyocites54.
FF reduced the IFNγ and IL-1β-induced cell proliferation of astrocytes in culture55.
| Reference | FF activity |
|---|---|
| Avis I, 200136 | Exposure of breast cancer cells to a 5-LO inhibitor up-regulated PPARα and PPARγ expression. Cells were growth inhibited when exposed to relevant PPAR agonists. |
| Chinetti G, 199837 | Macrophages activated by TNFα may suffer apoptosis by PPARα and PPARγ ligands. |
| Holland CM, 200413 | Treatment with FF significantly reduced proliferation and increased cell death in endometrial cancer cells, suggesting that altered expression of nuclear hormone receptors involved with fatty acid metabolism leads to deregulated cellular proliferation and apoptosis. PPARα was increased more than 4-folds in endometrial cancer cells. |
| Kubota T, 200516 | FF causes caspase-dependent apoptosis in human hepatocytes by inhibiting phosphorylation of Akt, in which PPARα is not involved. (Human hepatocytes) |
| Zak Z, 201038 | FF induces effective apoptosis in mantle cell lymphoma by inhibiting the TNFα/NF-kB signaling axis. |
| Wang J, 201139 | Combination of FF and cisplatin can enhance the role of cisplatin killing lung cancer A549 cells, which may be a result of up-regulating caspase-3 and down-regulating survivin. |
| Li T, 201440 | FF has anti-proliferation effects on breast cancer cell lines. Apoptosis was independent on the PPAR-α status with up-regulation of Bad, down-regulation of Bcl-xl, survivin and activation of caspase-3. FF produced cell cycle arrest at G0/G1 phase with down-regulation of cyclin D1, Cdk4 and up-regulation of p21, p27/Kip1. In vivo, FF slowed down tumor growth and induced apoptosis in the MDA-MB-231 xenograft mouse model. |
| Binello E, 201441 | FF showed anti-proliferative and pro-apoptotic effects on high-grade gliomas and anti-invasive effects on glioma stem cells. |
| Majeed Y, 201442 | In angiosarcoma forming endothelial cells FF induced apoptosis, reducing Bcl-2, survivin, Akt expression and Erk proteins. No effects on normal endothelial cells. |
| Reference | FF activities |
|---|---|
| Thuillier P, 200056 | Ligands for PPARα reduce skin cancer by 30% against skin tumor promotion. |
| Jiao H, 20027 | FF inhibits the growth of human HepG2 cells in a dose-related manner and oxidative stress was involved in this effect. |
| Holland CM, 200413 | Treatment with FF significantly reduced proliferation and increased cell death in endometrial cancer cells. |
| Gizard F, 200557 | FF inhibits vascular smooth vessel proliferation by inducing anti-oncogen p16. |
| Saidi SA, 200658 | The combination of FF and retinoic acid is a potent inhibitor of Ishikawa endometrial cancer cell growth in vitro. Cell cycle arrest is produced at G1/S. Cyclin D1 was down-regulated. These results could not be reproduced in vivo. |
| Panigraphy D, 200721 | FF suppresses tumor growth through angiogenesis inhibition. |
| Yokoyama Y, 200759 | Clofibric acid (a PPARα agonist) significantly suppressed the growth of OVCAR-3 tumors xenotransplanted s.c. and significantly prolongs the survival of mice with malignant ascites derived from DISS cells as compared with control. Microvessel density was diminished and apoptosis was also found. VEGF and PGE2 were also diminished. |
| Yamasaki D, 201160 | FF suppresses growth of the human hepatocellular carcinoma cell (Huh7) via PPARα-independent mechanisms and produces G1 arrest caused by the reduction of cyclin A and E2F1 and accumulation of the cyclin-dependent kinase inhibitor p27. This activity was not modified by PPARα antagonists. Inhibition of Akt phosphorylation by increased CTMP was also observed. |
| Chang NW, 201161 | FF highly suppresses the formation of squamous cell carcinoma in an oral-specific 4-nitroquinoline 1-oxide/ arecoline mouse model, decreases the tumor size, and increases the immunoreactivity of EGFR and COX2 in oral dysplasia, but decreases EGFR and COX2 expressions in SCC. These molecular events might be linked to the EGFR and COX2 regulatory pathways. |
| Huang J, 201362 | FF is capable to suppress B-cell lymphoma growth. This growth suppression is independent of angiogenesis inhibition. |
| Binello E, 201441 | FF shows anti-proliferative pro-apoptotic effects on high-grade gliomas and anti-invasive effects on glioma stem cells. |
| Li T, 201440 | FF has anti-proliferation effects on breast cancer cell lines. |
| Wang H, 201463 | FF can inhibit the growth and migration of human ovarian cancer cell SKOV3 in vitro, and to some extent induce apoptosis. |
| Liang H, 201464 | Synergistic inhibitory effects on cancer cell proliferation by simultaneous application of FF and budesonide. FF inhibited cell proliferation in both TP53 wild type and deficient lung cancer cells. The anti-proliferation effect of budesonide in TP53 wild type A549 cells was abolished in SK-MES-1 cells that do not have wild type TP53 protein. |
Antiproliferative activities are summarized in Figure 5.
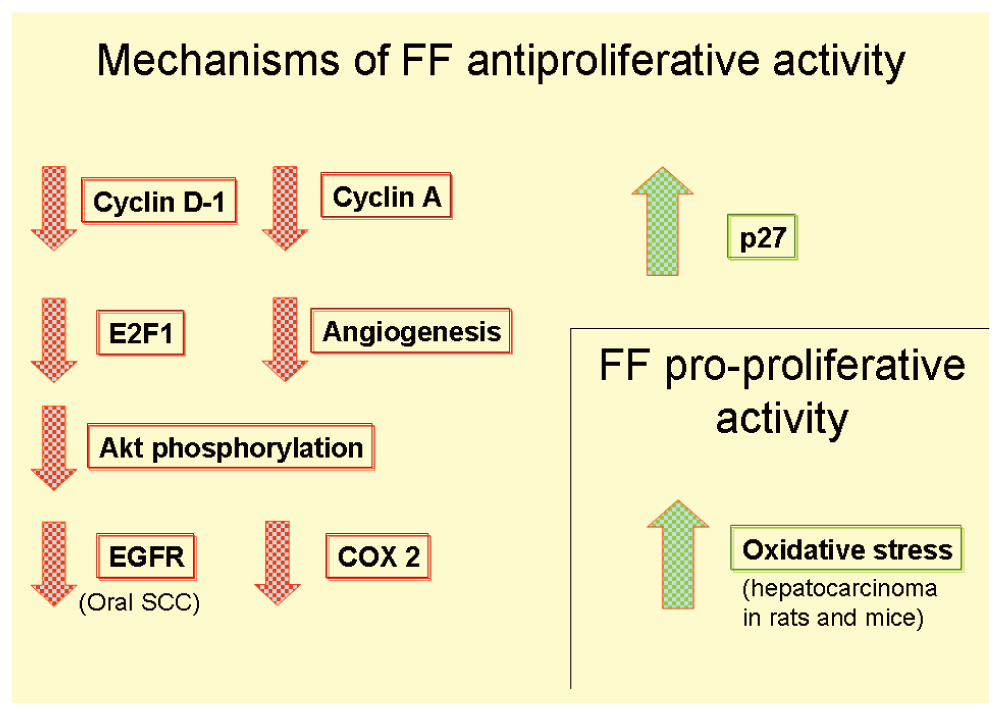
The work by Saidi et al.58 needs further discussion. The authors noticed that in Ishikawa endometrial cancer cells FF enhanced growth inhibition when ATRA was simultaneously used. ATRA by itself had no effect on growth. This is a logical finding because PPARα forms a heterodimer with RXR before binding DNA at the peroxisome proliferators responsive element. So this synergy between FF and ATRA regarding growth inhibition seemed a PPARα-dependent activity. Apoptosis also was increased with the combination of these drugs.
Paradoxically, RNAi inhibition of PPARα showed only a minor reduction in FF effect and ATRA combined with FF showed minor differences in growth inhibition with or without PPARα RNAi. After 48 hours of treatment the difference was approximately 40% less viability in cells treated with FF plus ATRA and no RNAi against those with RNAi. We hypothesize that RNAi inhibition of PPARα needs at least 48 hours to make the viability difference. So that growth inhibition seems, at least partially, as PPARα dependent.
They also found down-regulation of two genes: cyclin D1 (CCND1) and methionine adenosyltransferase 2 A (MAT2A), both are pro-growth genes58. High doses of FF up-regulated p21 (cyclin-dependent kinase inhibitor 1a) and TP53.
Unfortunately FF and FF plus ATRA showed no differences in tumor size and growing in vivo compared with control group receiving no drugs.
The work by Chang et al.61 suggests that FF may be useful for prevention of oral SCC because in an experimental setting FF was capable of reducing the incidence of tumors and also the progression from pre-neoplastic stage to SCC. FF at low doses lacked anti-tumor activity.
In spite of the known fact that glucocorticoids induce chemotherapy resistance in most of the solid tumors65, Liang et al.64 found that FF and budesonide had synergistic anti-proliferative effect on lung cancer cells with intact TP53.
Inflammation plays a very important role in carcinogenesis and tumor progression. NF-kB pathway is an essential actor of the pro-inflammatory and anti-apoptotic activity66–68.
NF-kB pathway increases angiogenesis, proliferation, anti-apoptosis, metastasis and inhibition of differentiation69.
FF has the capacity to down-regulate NF-kB activity according to evidences gathered in Table 4. Through this PPARα-dependent mechanism, FF exerts anti-inflammatory activity. Besides, it also has non PPARα-dependent anti-inflammatory activity through up-regulation of SHP (small heterodimer partner).
| Reference | FF activity |
|---|---|
| Staels B, 199870 | In aortic smooth-muscle cells PPARα ligands inhibit IL-1-induced production of IL-6 and expression of COX -2 as a result of PPARα down-regulation of NF-kB signaling. |
| Xu X, 200171 | FF and dexamethasone reduced NF-kB binding to its recognition site on the IL-6 promoter. FF reduced NF-kB binding to the vascular cell adhesion molecule-1 promoter. |
| Ogata T, 200472 | FF down-regulates NF-kB in myocardium of DOCA salt rats. IL-6, COX-2, VCAM-1 and MCP-1 were also down-regulated. |
| Okamoto H, 200518 | FF inhibits NF-kB signaling in rheumatoid synovial fibroblasts. |
| Yang TL, 200573 | FF reduce NF-kB in endothelial cells. This is a PPARα dependent activity. |
| Xu J, 200674 | FF reduces the signs of inflammation in murine astrocytes and down-regulated pro-inflammatory cytokines TNF-alpha, IL-1beta, and IL-6 by LPS-stimulated astrocytes. FF inhibited NF-kB DNA binding activity. |
| Li L, 201075 | FF attenuates tubulointerstitial fibrosis and inflammation through suppression of NF-kB and transforming growth factor-β1/Smad3 in diabetic nephropathy. |
| Zak Z, 201038 | FF induces effective apoptosis in mantle cell lymphoma by inhibiting the TNFα/NF-kB signaling axis. |
| Yang CS, 201376 | FF improved systemic inflammatory responses through the nuclear orphan receptor SHP (small heterodimer partner) and UCP2 (uncoupling protein 2). This is PPARα independent anti-inflammatory mechanism. |
| Schen W, 201477 | FF down-regulated NF-kB in endotoxin induced uveitis. All inflammatory factors like cytokine production, vessel density, vascular leukostasis and inflammatory cell infiltration was also down-regulated. |
| Binello E, Germano IM, 201478 | FF treatment decreased glioblastoma stem cell invasion in vitro. Treatment decreased the expression of NF-kB and cyclin D1 in a dose dependent and p53 independent manner. |
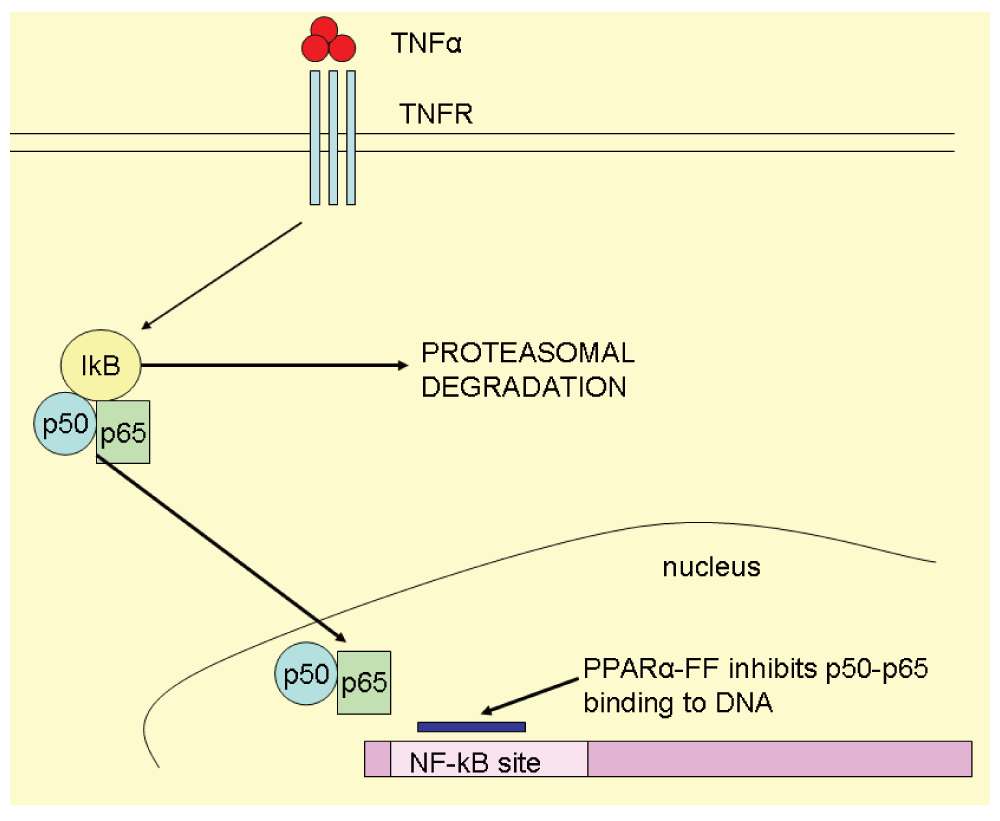
Evidences reported in Table 4 strongly support the FF anti-inflammatory activity mediated through NF-kB down-regulation and also PPARα independent mechanisms.
One of the proposed mechanisms of FF inhibiting NF-kB activity is depicted in Figure 6.
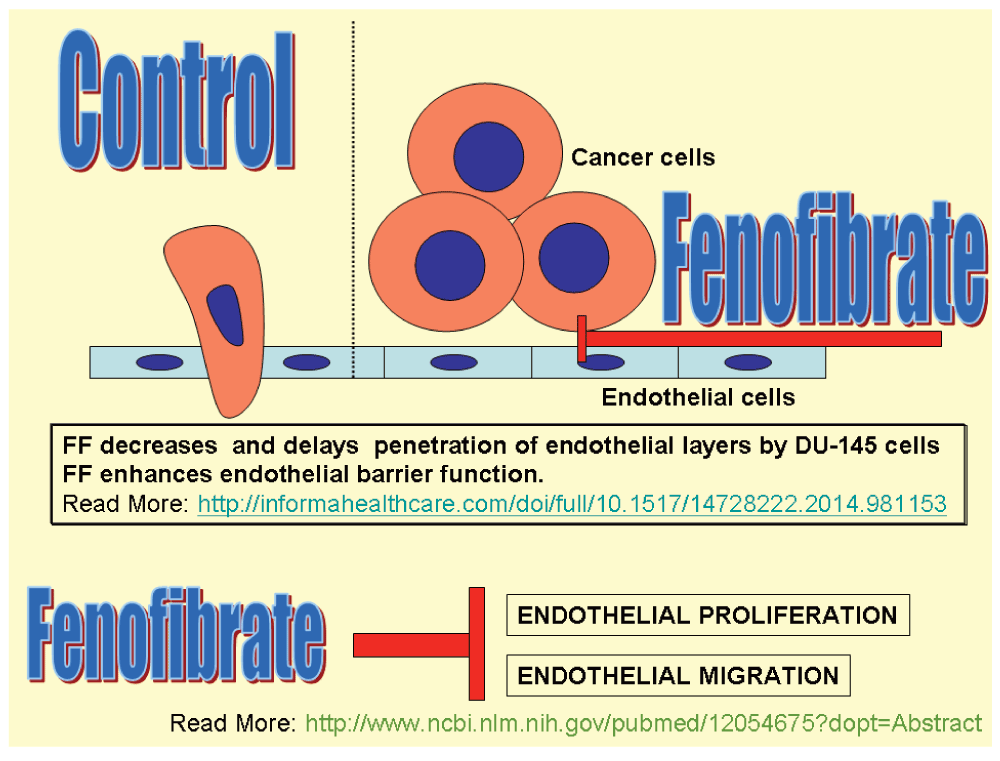
The research studies reported in Table 5 are evidence of down-regulation of Akt phosphorylation by FF, but Piwowarczyk et al.82 working with prostate cancer cells (DU-145) and endothelial cells (HUVEC) co-cultures found that FF increased levels of phosphorylated Akt in both HUVEC and DU-145 cells. They found that Akt phosphorylation was essential for FF increase of endothelial barrier (Figure 7).
| Reference | FF activities |
|---|---|
| Goetze S, 20026 | PPARα agonists decrease endothelial cell migration by down-regulating Akt. |
| Kubota T, 200516 | FF causes apoptosis in cultured human hepatocytes by inhibiting Akt. |
| Grabacka M, 200679 | PPARα activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. |
| Li R, 200780 | In cardiomyocytes FF and overexpression of PPARα inhibited endothelin-1 (ET-1)-induced phosphorylation of Akt and glycogen synthase kinase3β (GSK3β). |
| Yamasaki D, 201160 | FF suppresses growth of the human hepatocellular carcinoma cell via PPARα-independent mechanisms. It produces G1 arrest caused by the reduction of cyclin A and E2F1 and accumulation of the cyclin-dependent kinase inhibitor p27. FF also leads to suppression of Akt phosphorylation by increasing C-terminal modulator protein (CTMP), which inhibits Akt phosphorylation. FF inhibits proliferation by blocking Akt activation, and CTMP is one of the key players for this effect. |
| Majeed Y, 201442 | In angiosarcoma forming endothelial cells FF induced apoptosis, reducing Bcl-2, survivin, Akt expression and Erk proteins. No effects on normal endothelial cells were observed. |
| Kuno T, 201481 | FF can prevent the development of 4-NQO-induced proliferative lesions in the lung by modulating the insulin-IGF axis. FF significantly reduced the serum insulin and insulin-like growth factor (IGF)-1 levels, and decreased the immunohistochemical expression of IGF-1 receptor (IGF-1R), phosphorylated Akt, and phosphorylated Erk1/2 in lung adenocarcinomas. |
| Reference | FF activity |
|---|---|
| Keller BJ, 199283 | Clofibric acid and ciprofibrate inhibited stage 3 oxygen uptake in isolated mitochondria. |
| Zhou Z, 199984 | Fenofibrate, clofibrate, and ciprofibrate caused a direct dose-dependent depolarization of mitochondrial membrane potential which produced inhibition of oxydative phosphorilation and inhibition of ATP synthesis. Authors postulate that these are early events independent of PPARalfa activation. |
| Casas F, 200085 | FF or fasting suggests that the two treatments affect mitochondrial activity essentially by stimulating mitochondrial genome transcription, and increasing Cytochrome c oxidase activity. |
| Brunmair B, 200486 | Fenofibrate Impairs Rat Mitochondrial Function by Inhibition of Respiratory Complex I. |
| Nadanaciva S, 200787 | FF inhibits mitochondrial Complexes I, II+III, and V. |
| Wilk AM, 201288 and 2015157 | In glioblastoma FF is accumulated in the mitochondrial fraction, followed by an immediate impairment of mitochondrial respiration at the level of complex I of the electron transport chain. This mitochondrial action sensitizes tested glioblastoma cells to the PPARα-dependent metabolic switch from glycolysis to fatty acid β-oxidation. |
| Yang CS, 201376 | FF inhibits systemic inflammatory response through small heterodimer party (SHP) that acts through mitochondrial uncoupling protein 2 (UCP-2)*. |
Mitochondrial uncoupling proteins (UCP) are mitochondrial anion carrier proteins that separate oxidative phosphorylation from ATP synthesis with energy lost as heat and reduction of mitochondrial membrane potential89. The main function of UCP2 is the control of mitochondria-derived reactive oxygen species. PPARα modulates UCP2 expression90. Pecker et al. have demonstrated that UCP2 exerts control on proliferation: cells (embryonic fibroblast) where UCP2 expression was down-regulated grew faster than cells expressing UCP291. They also found that loss of UCP2 produced a metabolic change toward glucose metabolism, decreased fatty acid oxidation and increased proliferation.
Gamerdinger et al.166 found that FF and cyclooxygenase inhibitors like celecoxib (COX2 inhibitor) had a similar effect to cholesterol on cellular membranes, increasing it's rigidity and thickness. This conformational change of membranes produces changes in the functions of proteins embedded or spanning cell membranes, particularly in SERCA (sarcoplasmic reticulum Ca++ ATPase9) . (Li et al. 2004)167.
Youssef and Badr in 1998168 and Zungu et al.169 described the derangement of mitochondrial function due to peroxisome proliferator drugs. FF acts as an inducer of mitochondrial citrate synthase and NADH oxidase activity170 and Scatena et al.171 identified these mitochondrial changes in the respiratory chain consisting in inhibition of NADH cytochrome c reductase activity in a dose dependent manner. They also described that mitochondrial activities of FF were PPARα independent172,173.
Wilk et al.157 found that FF accumulated in mitochondria instead of the active form of the drug, the fenofibric acid, so that unchanged FF was the responsible of the activity on this organelle. This finding shows that FF and fenofibric acid may exhibit different pharmacologic activities (Figure 8), and explains the efforts done to find a suitable carrier to FF that would permit the arrival of this pharmaceutical to the tumor before being cleaved to FA174.
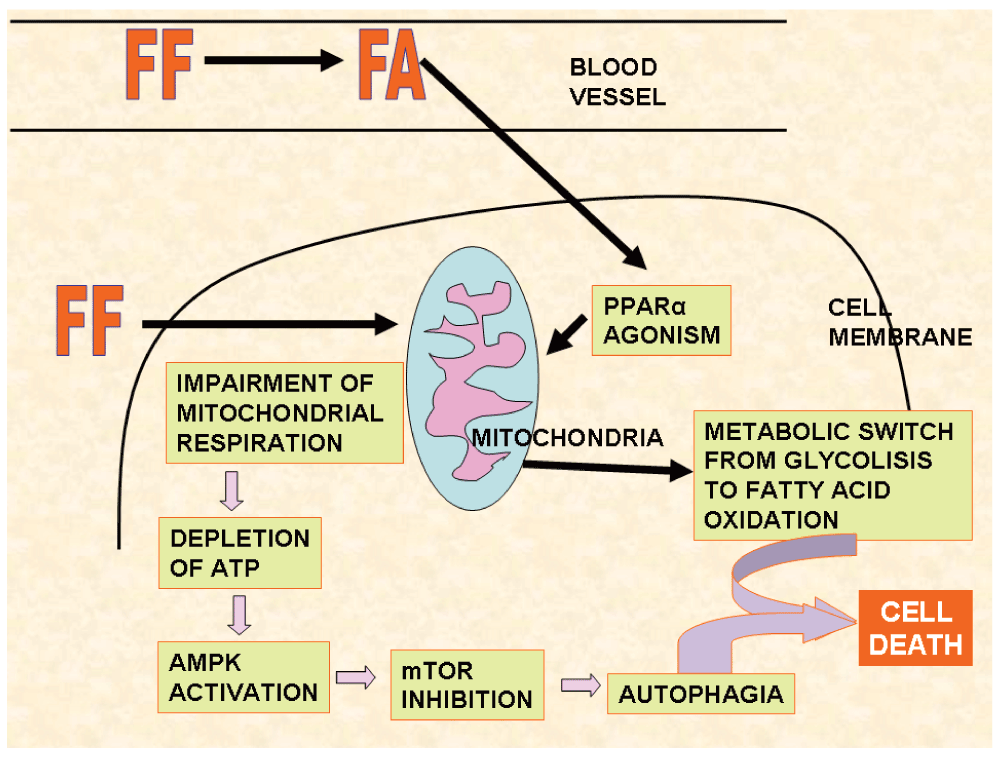
When FF is applied to the glioblastoma cell it accumulates in mitochondria and inhibits respiratory complex, depleting cellular ATP which activates AMPK, stimulating autophagia. The metabolic switch produced by PPARα activity of FF plus autophagia sensitizes cells towards death.
An important finding was that FF accumulates in the cell membrane fraction. FA does not174. This accumulation may be the explanation why FF increases Aβ42 (amyloid β peptide 42) production in mice brain175.
Evidence supports that FF decreases intracellular energy through inhibition of mitochondrial enzymes in a similar way to metformin. On a theoretical basis, we may assume that there might be synergism with metformin on this ground.
PPARα involvement with cancer metabolism has been extensively reviewed by Grabacka and Reiss92.
Another enzyme down-regulated by FF is FAS (fatty acid synthase)93 which is highly expressed in many cancer tissues. Fatty acid synthase (FAS) is a multicomplex enzyme that intervenes in endogenous synthesis of fatty acids and particularly palmitate. Abnormal fatty acid (FA) synthesis is one of the common features of many cancer cells and FAS has been identified as part of cancer controlling networks. Human cancers that over-express FAS, are usually associated with poor prognosis94–98.
The expression of adhesion molecules on the endothelial cell surface is critical for cells rolling in the vascular lumen to achieve tethering and adhesion to the vascular wall and eventually achieving diapedesis and colonization in the case of potentially metastatic cells or leukocyte recruitment to atherosclerotic lesions.
PPARα regulates gene expression of certain adhesion molecules in response to unsaturated fatty acids and fibric acid derivatives like FF. This control is achieved probably through inhibition of TNFα induced NF-kB activation99.
The research by Marchesi et al.99 that demonstrated reduction in adhesion molecules with FF treatment is important for two reasons:
1) It was performed in humans (10 hypertriglyceridemic patients).
2) The amount of reduction in fasting conditions (near 45% reduction for ICAM and around 33% reduction for VCAM levels).
Empen et al. (2003) described 10% reduction of E-selectin after six weeks treatment with FF, but found no major changes with VCAM-1 and ICAM-1 levels12.
Piwowarczyc et al.82 demonstrated a new FF effect: increased endothelial cell adhesion to the susbstratum and increased adhesion between endothelial cells by activation of focal adhesion kinase (FAK). These impedes cell diapedesis through the vessel wall, which is an important objective to decrease metastatic risk. Figure 7 depicts this activity.
The production of the metastatic cascade is a complex process in which there are many successive steps that we shall not analyze in depth in this review.
But for a better understanding lets remember the main steps106:
1) Primary tumor growth and angiogenesis.
2) Future metastatic cells free themselves from the primary tumor.
3) These cells degrade surrounding matrix.
4) Reach endothelium of vessels.
5) Enter blood vessels.
6) Circulate and survive in circulation.
7) Reach the target organ.
8) Attach to endothelial cells.
9) Migrate through vessel wall.
10) Start growth in the colonized site including angiogenesis.
11) Produce new metastatic cells.
12) Reinitiate the whole process.
Cell motility, invasion, angiogenesis and the function of connexins and adhesion molecules are an essential part of this cascade and have already been considered. In table 8 we describe only specific research work relative to metastasis and FF.
| Reference | FF activities |
|---|---|
| Marx N, 19993 | FF inhibits VCAM-1 transcription probably by inhibiting TNFα induced NF-kB activation. |
| Rival Y, 20025 | Fenofibric acid and FF reduced expression of VCAM-1 in endothelial cells and diminished migration of p65 fraction of NF-kB to the nucleus. |
| Marchesi S, 200399 | Reduce VCAM and ICAM in hypertriglyceridemic patients. |
| Gervois P, 2004100 | Suppression of IL-6-induced acute phase response gene expression after in vivo treatment with FF. Suppression of IL-6 reduces activation of STAT3 and c-Jun. |
| Lefebvre P, 2006101 | PPARα inhibits genes induced by NF-kB, such as VCAM-1, COX-2, and IL-6. |
| Ryan KE, 2007102 | FF decreased TNFα, IL-6 IL-1 beta VCAM and ICAM in obese glucose tolerant men after 6 weeks of treatment. |
| Rosenson RS, 2007103 | FF lowered fasting and postprandial VCAM-1 (-11%) and fasting and postprandial ICAM-1 (-15%) in patients with metabolic syndrome and hypertriglyceridemia. |
| Lee JW, 2007104 | FF repressed IFN-γ and IL-17 expression in isolated T cells. FF also repressed the expression of the genes encoding 3 chemokines, CXCL10, CCL2, and CCL20, and repressed CXCL10 gene promoter activity in tumor necrosis factor-α–treated HT-29 cells. |
| Li H, 2007105 | The E-selectin and ICAM-1 secreting could be inhibited by FF in endothelial cells. |
| Piwowarczyc K, 201482 | FF decreased cancer cell diapedesis by augmenting endothelial cell adhesion to the substratum. This was accompanied by the activation of ROS signaling, Akt and focal adhesion kinase (FAK) phosphorylation, in the absence of cytotoxic effects in endothelial cells. |
| Reference | FF activities |
|---|---|
| Grabacka M, 200414 | Inhibition of melanoma metastases by FF. |
| Grabacka M, 200679 | PPARα activation decreases migration, colony formation and metastatic potential of Human and mice melanoma cells in vitro via down-regulation of Akt and ERK ½. |
| Wybieralska E, 2011107 | Two parameters crucial for cancer cell metastatic potential, i.e. cell motility and gap junctional coupling, are inhibited by FF in DU 145 prostate cancer cells decreasing invasive potential. |
COX-2 is the rate-limiting enzyme in prostaglandin synthesis that catalyzes the production of prostaglandins and thromboxanes from arachidonic acid, and has been associated with growth regulation and carcinogenesis in many tumors. The COX2/PGE2 pathway may be considered a pro-tumor pathway at least in certain cancers where elevated levels of COX2 have been identified. Most colorectal carcinomas and many adenomas exhibit this elevation108,109. One of the postulated mechanisms by which COX2/PGE2 signaling stimulates cell growth is through the activation of β-catenin110. COX2 is implicated in breast cancer progression and invasiveness111,113. In stage III breast cancer, COX2 over-expression is an unfavorable prognostic sign, and according to Kim et al. gives ground for using COX2 inhibitor combinations112. Simeone et al. identified the pathway leading to increased invasiveness in breast cancer: COX2 /protein kinase C/interleukin-8/urokinase-type plasminogen activator pathway114. COX2 is also associated with angiogenesis and metastasis115.
Many cancers harbor increased COX2 activity including lung, colorectal, breast and squamous cell carcinoma of the upper digestive system116–118. COX2 down-regulation is an important issue in many cancers. We have been describing the action of FF on different pro-tumor proteins in an artificially separate manner, but many of these proteins share their activity in the pro-tumor evolution or are part of the same pathway. This is the case of NF-kB and COX2 in the progression towards cancer of Barret’s esophagus in which increased NF-kB activity is linked to increased IL-8 and COX2 expression119. As described above, FF is active against both: NF-kB and COX2.
The anticancer activities of FF are pleiotropic. Besides proliferation and angiogenesis down-regulation representing the main anti-tumoral effects, there are many others that will be described in the next three tables like ovarian aromatase inhibition, AMPK activation, IGF-I down-regulation, etc.
The IGF-1 receptor signaling system is a contributing factor in invasion, migration and proliferation of glioblastoma and became a legitimate target in the treatment of this pathology127. FF has experimentally shown to inhibit this system and decrease growth and invasion81,125,126. A sort of IGF-1 trap was designed by D'Ambrosio et al.128 that inhibited tumor growth in vivo and induced apoptosis.
These publications reported contrasting results: two of the showed that FF increased radiation sensitivity132,133 and one showed decreased radiation sensitivity131. As we shall see latter, PPARα agonists are tissue-specific and species-specific. This may explain the difference. In the first case the experiments were performed on HeLa cells and in the second and third studies, experiments were performed on squamous cell carcinoma cells. In all three cases human cells were used, so the difference may lay in tissue-specific behavior or may be due to the fact that in the second experiment the environment was particularly hypoxic.
Semaphorins are a large family of axon guidance molecules. They interact with their receptors, plexins and neuropilins, and play important roles in a growing list of diverse biological systems, including cancer progression and tumor angiogenesis. Some semaphorins can activate tumor progression and angiogenesis, while others may have the opposite effect.
There is abundant literature on semaphorins 3,4 and 7. Little is known about semaphorin-6B. It is known that there is an association between gastric cancer136, gliobastoma134 and certain breast cancers (MCF-7 breast adenocarcinoma cell line135) and semaphoring-6B but the exact nature of this association is still poorly understood.
PPARα agonists clofibrate and fenofibrate can down-regulate semaphorin-6B gene expression.
According to Ge136, inhibition of semaphoring-6B expression via RNA interference inhibited the migration, adhesion and invasion abilities of SGC-7901 gastric cancer cells in vitro.
SEMA6B transcript was down-regulated in two human glioblastoma cell lines (T98G and A172) when a prolonged treatment with ATRA was performed137.
The SEMA6B gen has a PPARα binding site in the promoter region so that the interrelation of PPARα and the gene effectively is present, and this interrelation is a negative one and probably exerts anti-tumoral effects.
In a large population study in Finland to assess the overall risk of prostate cancer in people taking cholesterol lowering medication, no decrease in risk was seen either with statins nor fibrates141. The statistics is significantly biased regarding fibrates: the population is too small (around 220 patients with prostate cancer receiving fibrates out of a total number of prostate cancer cases of 24.723).
FF was tested in different tumor cell lines. Table 16 summarizes this research.
HIV treatment with protease inhibitors has a frequent side effect: lipodystrophy syndrome. FF has been very effective in the treatment of this syndrome142. Nelfinavir and ritonavir are protease inhibitors which have shown many anti-cancer activities. We have postulated nelfinavir as a complementary off target treatment for cancer143. The association of FF with nelfinavir may prevent lipodystrophy, hypertriglyceridemia and elevation of lipoparticules144–146. There is no experimental proof of synergy of this interaction regarding cancer treatment, but both drugs share certain characteristics that make synergy a very plausible feature.
Nelfinavir is a proteasome inhibitor, and proteasome is the site where PPARα is dissembled, so nelfinavir should prolong PPARα’s life. Moreover nelfinavir and FF share the following activities: Akt inhibition, anti-angiogenesis, decreased proliferation, increased apoptosis, reduction of MMP2 and MMP9, and increased p21143.
Table 17 shows similarities and differences between nelfinavir and FF. The main purpose of this table is to illustrate the similarities and differences between this two drugs regarding cancer: nelfinavir down-regulates CDK-2 and FF down-regulates cyclin D-1 reinforcing cell cycle slowdown. Both are strong anti-angiogenic agents. FF also down-regulates COX2 which is an important step in decreasing angiogenesis.
| Reference | FF activities |
|---|---|
| Chen XR, 2007120 | In a brain damage experimental model performed in rats, Chen et al. demonstrated that FF improved neurological recovery by anti-inflammatory effects: decrease in iNOS, COX2 and MMP9 expression. |
| Varet J. 20038 | Reduced COX2 expression. |
| Ogata T, 200452 | In DOCA salt hypertensive rat model, FF reduced COX 2 expression, besides decreasing IL-6, VCAM-1 and MCP-1. |
| Chen YJ, 2008121 | FF reduced COX2 expression in kidney of diabetic rats. |
| Lee DL, 2011122 | Similar results as in Ogata T: in a DOCA salt hypertensive rat model FF reduced COX2 and IL-6expression. |
| Chang NW, 201161 | FF reduces COX2 expression in an oral SCC experimental model. |
| Panigraphy D, 200821 | Cox 2 was reduced in melanoma B16-F10 cell line when treated with FF. |
| Alvarez de Sotomayor M, 2007123 | FF decreased COX1 and COX2 in endothelium of resistance arteries in rats. |
| Ramanan S, 2008124 | Irradiating a murine microglial cell line with γ-rays led to increased expression of IL-1β and TNFα, Cox2. FF significantly decreased over-expression probably through NF-kB down-regulation. |
| Reference | FF activities |
|---|---|
| Urbanska K, 2008125 | In medulloblastoma cell lines, FF inhibits IGF-1 and growth by cell cycle arrest and induces apoptosis. |
| Drukala J, 2010126 | ROS accumulation and IGF-IR inhibition contribute to FF/PPARα mediated inhibition of glioma cell motility in vitro. |
| Kuno T, 201481 | FF exerts modulation of insulin-IGF-IGFR axis. |
| Reference | FF activities |
|---|---|
| Toda K, 2003129 | FF inhibits ovarian estrogen synthesis by suppressing the aromatase mRNA expression. Functional PPARα is indispensable for the inhibitory action of the agent in vivo. |
| Zhao H, 2013130 | FF down-regulates the expressions of androgen receptor (AR) and AR target genes and induces oxidative stress in the prostate cancer cell line LNCaP. |
| Reference | FF activities |
|---|---|
| Liu Z, 2012131 | FF can reduce radiation sensitivity by ROS scavenging via SOD induction in HeLa. SOD induction by FF is related with PPARα. |
| Ge Y, 2014132 | FF enhances radiosensitivity of esophageal squamous cell carcinoma by suppressing hypoxia-inducible factor-1α expression. |
| Liu J, 2014133 | FF enhances radiosensitivity of HNSCC by inducing arrest and apoptosis. |
| Collet P, 2004134 | Established that PPARα activators (clofibrate) diminished the expression of the human SEMA6B gene. Expression of SEMA6B gene in human glioblastoma T98G cells was down-regulated with clofibrate. |
| Murad H, 2006135 | Treatments with FF and troglitazone (a PPARγ ligand) strongly decreased the Sema6B mRNA. The drop in Sema6B mRNA level and in protein content was more important when the treatment combined the action of FF or troglitazone and 9-cis-retinoic acid. |
| Reference | FF activities |
|---|---|
| Murakami H, 2006138 | FF activates AMPK and increases eNOS phosphorylation in HUVEC cells without transcriptional activity. |
| Jiying W, 2012139 | Combined arsenic trioxide and FF exert a significant effect on epithelial-mesenchimal transformation of A549 cells, which may be related with the expression of E-cadherin and Snail. |
| Reference | PPARα agonists activities |
|---|---|
| Tuillier P, 200056 | PPARα agonists partially prevent tumor development in skin induced carcinogenesis. |
| Tanaka T, 2001140 | PPARα agonists prevent abnormal cript foci in induced colon cancer. |
| REFERENCE | TUMOR CELL LINE | SPECIE |
|---|---|---|
| Panigraphy, 200821 | MELANOMA | mice |
| Huang, 201362 | MELANOMA | human |
| Grabacka M, 200679 | MELANOMA | human and mouse |
| Wilk A, 201588 | GLIOBLASTOMA | mouse |
| Han DF, 2015158 | GLIOBLASTOMA | human |
| Binello E, 201441 | GLIOBLASTOMA | human |
| Wilk A, 2012156 | GLIOBLASTOMA | human |
| Drukala, 2010128 | GLIOBLASTOMA | human |
| Urbanska, 2008127 | MEDULLOBLASTOMA | mouse and human |
| Piwowarczyk, 201582 | PROSTATE CARCINOMA | human |
| Zhao H, 2013130 | PROSTATE CARCINOMA | human |
| Wybieralska E, 2011107 | PROSTATE CARCINOMA | human |
| Li T, 201440 | BREAST CANCER (triple negative) | human |
| Kuno T, 201481 | LUNG CANCER | mice |
| Liang H, 201464 | LUNG CANCER | human |
PPARα is highly expressed in certain cancer cells like endometrium26, prostate147, bladder148, certain breast cancer cell lines149, NSCLC151 and others.
In these cases, the administration of a PPARα agonist like FF increased apoptosis and decreased proliferation.
In human breast cancer cell lines MCF-7 and MDA-MB-231, PPARα was overexpressed but use of agonists of this receptor increased proliferation149.
In stark contrast, Li et al.151 tested FF in 12 breast cancer cell lines (including MDA-MB-231) and in all of them FF was effective as proliferation inhibitor. This effectiveness was independent of PPARα expression but was linked to triple negative condition. Paradoxically MDA-MB-231 was the more sensitive to inhibition of proliferation by FF treatment.
In human colon cancer tissues PPARα is underexpressed when compared with normal tissues150. Using PPARα ligands in APCMin/+ mice to evaluate polyp formation, those treated showed decreased number of polyps and decreased size. We may conclude that overexpression or under-expression of PPARα in cancer tissues is not an indicator of future response to FF or other PPARα agonists.
Another issue to consider is the species-specific response to PPARα agonists: for instance, FF induces hepatocarcinogenesis in rodents but not in humans, insulin resistance in mouse but not in humans, oxidative stress in mouse heart but not in human heart. Human liver has a lower expression of PPARα than rodents152 (The differences of PPARα in human liver has been extensively described in the review by Roberts153).
There are important differences between rat and human hepatic cells. Vanden Heuvel et al.154 studied the gene expression differences between rat hepatoma cells (FaO) and human hepatocarcinoma cells (HepG2) when treated with a PPARα agonist like WY14643. A large number of kinases and phosphatases were affected in FaO and not in HepG2 cells. Many of them were implicated in cell cycle control and growth signaling like JAK1, JAK2, GSK3α and MKP-1.
Rat peroxisomes contain urate oxidase which is absent in human peroxisomes155.
Acyl Co A oxidase (ACO) is a key enzyme in peroxisomes and according to Roberts et al.153 there are differences between human and rat ACO: the promoter for human ACO has a different sequence and activity from rats ACO.
PPARα agonists like FF are tissue-specific. They may increase VEGF and angiogenesis in retina22,23 and the opposite in tumor cells20,21.
Thus this species- and tissue-specificity suggests that we should be cautious when interpreting the results of many of the published investigations. PPARα agonists research results obtained in rodents should not be taken for granted in humans.
The mechanism of FF anticancer activity may differ in different tumors: in mantle cell lymphoma it seems to induce apoptosis by inhibiting the TNFα/NF-kB axis38 while in triple negative breast cancer it requires activation of NF-kB in order to induce apoptosis40.
PPARα agonists like FF are actively investigated as anti-cancer drugs, but paradoxically, PPARα inhibitors may also work against cancer. This is the case of renal cell carcinoma45 where an inhibitor of this receptor produced cell cycle arrest and apoptosis.
After these necessary clarifying precautions, we must consider in detail the hard evidence collected in the medical literature that gives ground for FF as a complementary adjunct pharmaceutical in cancer therapy.
Phanigraphy et al.21 tested 19 human tumor cell lines in vitro and found that all expressed PPARα in tumor cells and endothelium. There were differences regarding levels of PPARα. Fibrates and particularly FF at clinically achievable concentrations showed capacity to inhibit proliferation in these cell lines that included highly malignant ones like melanoma and Lewis lung carcinoma cell lines. FF inhibited 95% of bovine capillary endothelial cells proliferation and migration. There was no inhibition on normal fibroblasts growth. In glioblastoma cells FF reduced VEGF secretion by 50% and increased TSP-1 expression by 3- to 4- fold in a fibrosarcoma cell line. The results of these experiments show that FF exerts anti-proliferative and anti-angiogenic activities in tumor cells in vitro at clinically achievable concentrations. Similar results were observed in subcutaneously implanted tumors in mice. The seminal work of Panigraphy et al. concludes that anti-proliferative and anti-angiogenic properties of FF are PPARα activation dependent.
Glioblastoma treatment has already introduced FF as part of different anti-angiogenic schedules, but FF may have also other anti-tumoral effects in this disease besides anti-angiogenesis, particularly apoptosis156–158.
Endometrial carcinoma usually overexpresses PPARα. When PPARα expression is reduced with siRNA in vitro, cellular proliferation decreased substantially and showed a small increase in apoptosis in Ishikawa cells and HEC-1A cells27. Both cell lines reduced VEGF levels when they were treated with FF. Reduction in VEGF after FF treatment showed differences between the two cell lines: HEC-1A showed potentiation of inhibition of VEGF when an RXR ligand was added; Ishikawa cells did not. This reinforces the concept that there may appear important differences with FF treatment according to the tumor type.
FF also uses the AMPK pathway to produce its anti-tumoral effects159. This is a PPARα independent action that has been demonstrated in human oral squamous cell carcinoma (OSCC) where FF inhibited cell migration and invasion and reduced expression of MMP 1, 2, 7 and 9. LKB1 and AMPK were up-regulated after FF treatment. When AMPK was inhibited (with protein C) the anti-invasive effect was significantly reduced.
Metformin is another activator of the AMPK pathway. It has not been tested with FF but it is quite possible that there might be synergy in this activity. Another coincidence between metformin and FF is the decrease in cellular energy production.
The molecular mechanism of many of the FF anti-cancer activities is known since 2006160 and can be summarized as inhibition of COX2 and VEGF at the transcription level by interfering with AP-1 binding to DNA and decreased expression of NF-kB. In susceptible cells there is a negative cross talk between PPARα and AP-1160.
Another mechanism postulated in anti-cancer activity is the disruption of the tumor-host stroma symbiosis due to anti-angiogenesis and anti-inflammatory activity161.
Finally we have to mention that FF effectively reduces nuclear SREBP-2 but not SREBP-1162–165. Nelfinavir and PUFA inhibit mature SREB-1 which is a transcription factor that promotes FAS synthesis, so these products may complement FF anti-cancer activity.
As observed in the tables, FF exerts polyvalent anti-cancer activities that deserve further research in the clinical setting.
FF is a PPARα agonist drug developed for treatment of elevated triglycerides and LDL cholesterol reducing cardiovascular risk that may be repurposed to be used in cancer due to its anti-angiogenic, anti-inflammatory, anti-proliferative, anti-metastatic, anti-adhesive, anti-invasive and pro-apoptotic activities in certain cancers.
FF has already been incorporated in anti-angiogenic protocols for the treatment of glioblastoma. Probably in the future it will form part of protocols based on repurposed drugs directed to inhibit angiogenenesis like celecoxib, nelfinavir, and metformin.
Colorectal and prostate cancer seem good candidates for these therapies.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)