Keywords
LytSR, Histidine kinase, Response Regulator, Phosphotransfer, Staphylococcus Aureus
LytSR, Histidine kinase, Response Regulator, Phosphotransfer, Staphylococcus Aureus
The major differences between version 1 and this version of the manuscript are as follows:
See the authors' detailed response to the review by Kenneth W Bayles
See the authors' detailed response to the review by Taeok Bae
The molecular basis of signal transduction by LytSR is unknown. We show that LytS has kinase and phosphatase activity. LytR undergoes rapid phosphorylation by acetyl phosphate. Activity of LytR is regulated either through LytS or acetyl phosphate. LytSR is at the interface of two regulatory pathways that respond to excess glucose metabolism and cell membrane electrical potential, respectively.
The two-component system LytSR of Staphylococcus aureus is reported to function as a sense-response system for detecting subtle changes in the electrical potential of the cell membrane. It is also involved in adaptation of S. aureus to cationic antimicrobial peptides (CAMPs)1–3. CAMPs are bactericidal agents released by the host innate immune system during colonization by S. aureus4,5. Their mechanism of action is believed to involve perturbation of cell membranes which, in turn, alters the electrical potential of the cell membrane3,6. The function of LytSR in the cell is reported as a regulator of cell wall lysis during programmed cell death and biofilm formation7–12.
A typical two component system (TCS) consists of a membrane bound histidine kinase (HK) which intercepts an environmental cue and through an act of auto-phosphorylation transduces the signal intracellularly. The response to the cue is mediated through a phosphotransfer process whereby a second protein, in response to a regulator protein, receives the phosphoryl group from the cognate histidine kinase (HK) at a conserved aspartate residue. The response regulator (RR) protein is often a transcription factor and in some cases an enzyme13. LytSR is comprised of the membrane-bound sensor HK LytS and the RR protein LytR. LytS belongs to a family of bacterial receptor proteins that contain five transmembrane-spanning domains. LytR is a transcription factor that falls into a novel family of proteins that contain non-helix-turn-helix DNA-binding domains, known as LytTRs14,15.
LytR regulates lrgAB operon in response to alterations in the electrical potential of the cell membrane16. The lrgAB operon together with cidABC operon are involved in the control of programmed cell death and lysis during biofilm formation12,17,18; the gene products of the cid operon enhance murein hydrolysis activity and antibiotic tolerance19 while the lrg genes inhibit these processes9. Interestingly, both operons were also shown to be induced by carbohydrate metabolism16 and proposed to be regulated through a cidR-dependent signaling pathway1. The cidR gene encodes a LysR-type transcription factor which has been proposed to be activated by accumulation of acetate during metabolism of excess glucose by S. aureus at logarithmic growth20,21. Recent work by Sharma-Kuinkel et al.2 demonstrated that lrgAB is instead regulated only through LytR, either in response to carbohydrate metabolism (e.g. excess of glucose) or as a result of disruption of cell membrane electrical potential. However, the molecular basis for this observation remained obscure.
To examine the signal transduction mechanism of LytSR and probe its involvement in the regulation of lrgAB in response to carbohydrate metabolism as a result of a phosphorylation-induced activation of LytR by acetyl phosphate, we cloned and purified LytS and LytR and investigated the autokinase activity of LytS, the kinetics of the phosphotransfer between LytS and LytR, and the kinetics of phosphorylation of LytR by acetyl phosphate. Our study shows that LytSR is capable of mediating signaling either through LytS in response to cell membrane electrical potential or through LytR in response to carbohydrate metabolism. Furthermore, phosphorylation-induced activation of LytR by either LytS or acetyl phosphate is likely to involve dimerization of LytR at the receiver domain.
Chemicals and antibiotics were purchased from Sigma (Oakville, Canada) or Thermo-Fisher (Whitby, Canada), unless otherwise stated. Chromatography media and columns were purchased from GE Healthcare (Quebec, Canada). Growth media were purchased from Fisher. Escherichia coli strains, NovaBlue and BL21 (DE3), and cloning and expression plasmids were purchased from EMD4 Biosciences (New Jersey, USA). The pGEX-4T vector was purchased from GE Healthcare (Quebec, Canada). Restriction enzymes were obtained from New England Biolabs (Pickering, Canada) or Thermo-Fisher. The [γ-32P] Adenosine triphosphate (ATP) was purchased from Perkin Elmer LAS Canada Inc. (Toronto, Canada) or GE Healthcare. The Proteo Extract All-in-One Trypsin Digestion Kit was purchased from EMD4 Bioscience. The genome of the S. aureus strain Mu50 was obtained from Cedarlane (Burlington, Canada).
The gene sequence of lytS (SAV0260, as per gene numbering in S. aureus Mu50 strain) encoding the cytoplasmic region of the protein (amino acid residues 355–584) was amplified from S. aureus Mu50 genome using the primers: Dir 5'- CACCGCAGAAGGATTGGCAAAT-3' and Rev 5'-TTATTCCTCCTCTTG TCTTT CA-3'. To enable directional cloning, the forward primer contained a specific 4 base pair (bp) sequence (italicized) at the 5' end of the primer. The 701 bp lytS gene was amplified using Phusion High-Fidelity DNA polymerase with the following PCR conditions: initial denaturation at 98°C for 30 s, followed by 30 cycles at 98°C at 10 s, annealing at 61°C for 20 s, extension at 72°C for 20 s and final extension at 72°C for 10 min. The blunt ended amplicon was then ligated to pET151/D-TOPO vector using the Champion™ pET Directional TOPO® Expression Kit. The ligated pET151/D-TOPO::lytS construct was used to transform E. coli NovaBlue cells for further amplification. The correct sequence of lytS was confirmed by DNA sequencing (The Centre for Applied Genomics, the Hospital for Sick Children, Toronto, Canada. The pET151/D-TOPO::lytS plasmid was used to transform the E. coli BL21 (DE3) to facilitate protein expression. This cloning strategy resulted in introduction of an NH2-terminous 6xHis tag upstream of lytS.
E. coli BL21(DE3) cells carrying the construct pET151/D-TOPO::lytS were used to inoculate 5 mL of Luria Bertani (LB) medium supplemented with 100 µg/mL final concentration of ampicillin and allowed to grow overnight at 37°C by shaking. An aliquot of 1 mL of the overnight cell culture was used to inoculate 1 L of Terrific Broth (TB) medium supplemented with 100 µg/mL of ampicillin. Cells were allowed to grow at 37°C by shaking at 200 rpm until the cell culture reached an optical density of 0.6 to 0.8 absorbance units at 600 nm (OD600nm). At this point, the cell culture was cooled to 4°C and protein production initiated by adding isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) at a final concentration of 0.1 mM. Cell culture was shaken at 200 rpm at 18°C for 12 hrs. Cells were harvested by centrifugation at 3,300 × g for 20 min.
For isolation of His-LytS, the cell pellet was resuspended in 50 mM sodium phosphate pH 7.5, supplemented with 300 mM NaCl and 10 mM Imidazole. The cellular content was liberated by sonication while cooling on ice for 10 min (10s on/15s off) and cell debris was removed by centrifugation at 18,000 × g for 1 h at 4°C. Purification of His-LytS was carried out by loading the supernatant onto a self-assembled affinity column (Generon, UK) packed with 8 mL of Ni-NTA resin. All purification steps were carried out at 4°C using the AktÄ purifier 10 (GE Healthcare). The unbound protein was removed by washing with buffer for three column volumes (CV). The protein of interest was eluted using a step gradient of 10%, 40%, 70% and 100% of 300 mM imidazole in 50 mM sodium phosphate pH 7.5 buffer, supplemented with 300 mM NaCl, at a flow rate of 1.5 mL/min in three CV. Fractions of 5 mL containing the protein were concentrated using Amicon Ultra-10K concentrator (Millipore) followed by dialysis into the storage buffer: 50 mM Tris pH 7.5, supplemented with 150 mM NaCl and 5 mM MgCl2. The homogeneity of protein was assessed by loading samples onto a 15% sodium dodecyl sulphate-polyacrimide gel electrophoresis (SDS-PAGE) apparatus and staining the gel with Coomassie blue.
The gene sequence of lytR (SAV0261) was amplified from the S. aureus Mu50 genome using the primers: Dir 5'-GGAATTCCATATGAAAGCATTAATCATAGATGATG-3' and Rev 5'-CGGAATTCTTAT TAAAGTAATCCTA TCGACG-3'. The primers were designed to introduce the NdeI and EcoRI restriction sites (italicized sequences) at 5' and 3' of lytR, respectively. Amplification of the 741 bp lytR gene was carried out using Phusion® High-Fidelity DNA polymerase following these conditions: initial denaturation at 98°C for 30 s, followed by 30 cycles at 98°C at 10 s, annealing at 62°C for 20 s, extension at 72°C for 20 s and final extension at 72°C for 10 min. The resulting amplicon was purified and together with the host vector, pET26b, was digested with NdeI and EcoRI. The digestion products were gel purified from 1% agarose gel using the QIAquick Gel Extraction Kit (Qiagen) and subjected to ligation using T4 DNA ligase (NOVAGEN). The subsequent construct was referred to as pET26b::lytR and was used to transform E. coli NovaBlue cells for further amplification. The correct sequence of the lytR gene was confirmed by DNA sequencing (The Centre for Applied Genomics, the Hospital for Sick Children, Toronto, Canada). The pET26b::lytR plasmid was used to transform the E. coli BL21 (DE3) expression cells. This cloning strategy resulted in introduction of no tags or additional amino acids to LytR.
To clone the receiver domain of the LytR protein, LytRN (residues 1-134), a stop codon after the 134th amino acid residue was introduced using the QuikChange® Site-Directed Mutagenesis method (Thermo Fisher). The process of site directed mutagenesis was carried out using the Pfu Turbo® DNA polymerase and the following mutagenic primers: 5'-GCGAATGATATGTCGTAGAATTTTGATCAAAGC-3' and 3'-GCTTTGATCAAAATTCTA CGACATATCATTCGC-5' (mutated nucleotides are italicized). The construct pET26b::lytR was used as the template. The amplified mutagenic construct, pET26b::lytRN was treated with the restriction endonuclease DpnI and used to transform E. coli NovaBlue cells. Successful insertion of the stop codon was confirmed by DNA sequencing (The Centre for Applied Genomics, the Hospital for Sick Children, Toronto, Canada) and the pET26b::lytRN vector was used to transform E. coli BL21 (DE3).
The following primer set was used to clone the full length lytR into pGEX-4T-1 to enable fusion of the protein to the C-terminal of GST, Dir 5'-AGTCGGGATCCATGAAAGCATTAATCATA GATG-3' and Rev 5'-CG GAATTCTTATTAAAGTAATCCTATCG ACG-3'. The primers were designed to introduce BamHI and EcoRI restriction sites (italicized sequences) at 5' and 3' of lytR ends respectively, to enable cloning.
The Asp-53 residue of LytR was mutated to an Ala residue using QuikChange® Site-Directed Mutagenesis (Thermo Fisher). Briefly, the process of site directed mutagenesis was carried out using the Pfu Turbo® DNA polymerase with the designed mutagenic primers: 5'-AC ATTATATTTTTAGCTGTCAATTTAATGG-3' and 3'-CCATTAAATTGACAGCTAAAA AT ATAATGTC- 5' (mutated nucleotides are italicized). The construct pGEX-4T1::lytR was used as a template. The amplified mutagenic construct, referred to as pGEX-4T::lytRAsp53Ala, was treated with the restriction endonuclease DpnI and used to transform E. coli NovaBlue. Successful mutation of Asp to Ala was confirmed by DNA sequencing (The Centre for Applied Genomics, the Hospital for Sick Children, Toronto, Canada). The pGEX-4T::lytRAsp53Ala plasmid was used to transform the E. coli BL21 (DE3).
In general, E. coli BL21 (DE3) carrying the appropriate plasmid was used to inoculate 5 mL of LB in the presence of 50 µg/mL of kanamycin. An aliquot of 1 mL of the overnight cell culture was used to inoculate 1 L of TB supplemented with 50 µg/mL of kanamycin. The cells were allowed to grow to OD600nm = 0.6–0.8 with shaking at 200 rpm at 37°C. Once desired growth was achieved the media was cooled to 4°C and protein production was induced by adding IPTG to a final concentration of 0.1 mM. Cell culture was allowed to shake for an additional 12 h at 18°C. Cells were harvested by centrifugation at 3,300 × g for 20 min.
To isolate LytR the method of protein precipitation by ammonium sulfate was employed. All purifications steps were carried out at 4°C. Cell pellet was suspended in 1:10 (w/v) of 50 mM Tris, pH 8.0, 100 mM NaCl, 5 mM MgCl2 supplemented with 10% glycerol. The cellular content was liberated by sonication and cell debris was removed by centrifugation at 18,000 × g for 1 h at 4°C. Total volume of supernatant was adjusted to be 50 mL (for 5 g cell) and 2.67 g of ammonium sulfate was added gently while stirring, to achieve saturation of 10%. The solution was incubated while stirring at 4°C for 30 min. The precipitated protein was collected by centrifugation at 3,300 × g for 5 min. The protein pellet was dissolved in 10 mL of 50 mM Tris, pH 8.0, 100 mM NaCl, 5 mM MgCl2 supplemented with 10% glycerol. The purity of the protein was evaluated by 15% SDS-PAGE stained with Coomassie blue. The protein solution was dialysed to remove ammonium sulfate.
E. coli BL21 (DE3) harboring pET26b::lytRN was used to inoculate 5 mL of LB in the presence of 50 µg/mL kanamycin. An aliquot of 1 mL of the overnight cell culture was used to inoculate 1 L of TB supplemented with 50 µg/mL of kanamycin. The cells were allowed to grow to an OD600nm = 0.6–0.8 while shaking at 200 rpm at 37°C. The cell culture was cooled to 4°C and protein production initiated by addition of 0.5 mM IPTG. The cell culture was allowed to shake for an additional 12 h at 25°C for. Cells were harvested by centrifugation at 3,300 × g for 20 min.
To isolate LytRN, cell pellet was suspended in 1:10 (w/v) of 20 mM Tris pH 7.5, supplemented with 5 mM MgCl2. The cellular content was liberated by sonication and cell debris was removed by centrifugation at 18,000 × g for 1 h at 4°C. The supernatant was loaded onto a DEAE-Sepharose™ column (GE Healthcare) and mounted into the AktÄ purifier 10. The protein was eluted over ten column volumes in a linear gradient of 20–500 mM Tris (pH 7.5) (supplemented with 5 mM MgCl2) at a flow rate of 3 mL/min. Elution fractions containing protein were pooled together and concentrated by centrifugation using Amicon Ultra-3K concentrator (Millipore) to a final volume of 5 mL. The protein was loaded onto a HiPrep 26/60 Sephacryl S-200 HR gel-filtration column (GE Healthcare). The homogeneity of the protein was determined using 18% SDS-PAGE.
E. coli BL21 (DE3) carrying the desired plasmid were used to inoculate 5 mL of LB in the presence of 100 µg/mL ampicillin. An aliquot of 1 mL of the overnight grown seed culture was used to inoculate 1 L of TB supplemented with 100 µg/mL of ampicillin. Cells were allowed to grow to an OD600nm = 0.6–0.8 at 37°C while shaking at 200 rpm. Then the cell culture was cooled to 4°C and protein production was initiated by addition of 0.5 mM IPTG and incubated further at 18°C for 12 h. The cells were harvested by centrifugation at 3,300 × g for 20 min.
For purification of the wild type or mutant GST-LytR protein, the cell pellet was suspended in 1:10 (w/v) of 50 mM Tris pH 7.5, supplemented with 100 mM NaCl and 5 mM MgCl2. The cellular content was liberated by sonication and cell debris was removed by centrifugation at 18,000 × g for 1 hour at 4°C. Purification was carried out by loading the supernatant onto a self-assembled affinity column packed with 5 mL of GST affinity resin (Generon). The protein of interest was eluted at a flow rate of 1.5 mL/min with 10 mM reduced glutathione in 50 mM Tris-HCl pH 8.0 buffer over three CV. Fractions containing the protein were concentrated using Amicon Ultra-10K concentrator (Millipore) followed by dialysis to exchange the buffer into 50 mM Tris pH 7.5, supplemented with 100 mM NaCl and 5 mM MgCl2. The homogeneity of protein was assessed by 12.5% SDS-PAGE stained with Coomassie blue.
The identities of the all proteins isolated in this study were confirmed by cutting the protein band from the SDS-PAGE gel, subjecting this to trypsin digestion and submitting the digest for mass spectrometry analysis. The molecular mass of the purified proteins was determined by electrospray ionization mass spectrometry (ESI-MS). All the mass spectrometry analyses were done at the Advanced Protein Technology Centre, Hospital for Sick Children (Toronto, Canada).
His-LytS at 5 µM was equilibrated in phosphorylation buffer (PB: 50 mM Tris, pH 7.4, 50 mM KCl, 5 mM MgCl2) supplemented with 10 mM CaCl2. The phosphorylation reaction was initiated by the addition of [γ-32P]-ATP (10 Ci/mmol) mixed with cold ATP to prepare reaction mixtures at different ATP final concentrations. Samples were incubated at room temperature. The reaction was stopped at different time intervals by adding 5 × SDS sample buffer (125 mM Tris, pH 6.8, 2.5% SDS, 25% glycerol, 100 mM dithiothretiol (DTT), 0.0025% bromophenol blue). Samples were loaded onto a 15% SDS-PAGE. The SDS-PAGE gels were washed in water containing 2% (v/v) glycerol and were exposed to a phosphor screen (GE Healthcare) overnight and imaged using a Typhoon Trio+ imager (GE Healthcare). The radioactive gels were stained by Coomassie blue dye to analyze protein content of the samples.
Each time-dependent phosphorylation experiment was repeated twice. The progress of the reaction was assessed by analyzing the phosphor-image of the radioactive gels using NIH ImageJ software (Version 1.45s) (freely available at http://imagej.nih.gov/ij/download.html). The progress curves were fitted to the first-order rate Equation 1, where I is the band intensity quantified by NIH ImageJ, kobs is the observed rate constant, t is the incubation time and A is the proportionality constant relating intensity with concentration of phosphorylated His-LytS.
I = A×{1 – exp(–kobs × t)} (1)
The data were fitted using Erithacus GraFit software (version 5.0.10) (available at http://www.erithacus.com/grafit/Software_Updates.htm). The observed phosphorylation rate constant was calculated for each ATP concentration and was plotted against each ATP concentration to determine the rate constant of autophosphorylation of LytS using the equation (2), where kobs is the observed rate constant measured at each ATP concentration, k is the autophosphorylation rate constant for LytS, Ks, is the dissociation constant of ATP and [S] the ATP concentration.
To study the effect of salt ions on the phosphorylation of His-LytS, we looked at the effect of K+ and Ca2+. His-LytS at 3 µM was equilibrated in the phosphorylation buffer (PB: 50 mM Tris, pH 7.4, 5 mM MgCl2) with varying concentrations of either KCl or CaCl2. The phosphorylation reaction was initiated by the addition of [γ-32P]-ATP (10 Ci/mmol) to a final concentration of 20 µM. The reaction was incubated for 90 min at RT then quenched by adding 5 × SDS sample buffer. Samples were loaded onto a 15% SDS-PAGE. The radioactive gels were washed in water containing 2% (v/v) glycerol and gels were exposed to a phosphor screen (GE Healthcare) overnight. The screen was imaged using a Typhoon Trio+ imager (GE Healthcare).
To investigate the stability of the phosphorylated state of LytS, His-LytS at 10 µM was allowed to undergo autophosphorylation for 90 min by adding [γ-32P]-ATP (10 Ci/mmol) to a final concentration of 20 µM. Excess [γ-32P]-ATP was removed by desalting using the Zeba Spin Desalting column (Pierce, Thermo Scientific) equilibrated with PB. The reaction mixture was further incubated at room temperature and aliquots were removed at different time intervals and quenched by adding 5 × SDS sample buffer. Samples were loaded onto 15% SDS-PAGE and the gel was analyzed as described above.
The ability of LytS to phosphorylate LytR was examined as described earlier22. Briefly, His-LytS at 15 µM was phosphorylated for 90 min. Excess [γ-32P]-ATP was removed by desalting using the Zeba Spin Desalting column which was equilibrated with PB. Phosphorylated His-LytS (4 µM) was incubated with GST-LytR (10 µM) in the PB buffer at room temperature. Aliquots of 10 µL were removed at different time intervals and quenched by adding 10 µL of 5 × SDS-PAGE sample buffer. Samples were loaded onto a 15% SDS-PAGE. The radioactive gels were washed with water containing 2% (v/v) glycerol and gels were exposed to phosphor screen (GE Healthcare) overnight and imaged using a Typhoon Trio+ imager (GE Healthcare). The radioactive gels were stained by Coomassie blue dye. The phosphor images of the radioactive gels were quantified using NIH ImageJ software (Version 1.45s). Similar experiments were carried out using GST-LytR-D53A mutant and LytRN.
To investigate phosphorylation of LytR by small molecule phosphate donors, lithium potassium acetyl phosphate was used as described earlier22. Briefly, LytR at 10 µM or LytRN at 20 µM was equilibrated in PB20 buffer (PB: 50 mM Tris, pH 7.4, 50 mM KCl, 20 mM MgCl2) and phosphorylation was initiated by addition of lithium potassium acetyl phosphate to a final concentration of 50 mM. The reaction mixture was incubated at 37°C and 15 µL aliquots were removed and quenched by adding 5 × SDS sample buffer. The phosphorylated species were separated from unphosphorylated species using a 15% SDS-PAGE containing Acrylamide-pendant Phos-tag™ AAL-107 at 50 µmol/L (Wako chemical USA, inc., Cedarlane)23. The gels were stained by Coomassie blue dye. Band intensities of the phosphorylated species were quantified using NIH ImageJ and plotted against incubation time. These data were fitted to Equation 1 using Erithacus GraFit software (version 5.0.10) to determine the phosphorylation rate constants of LytR and LytRN by acetyl phosphate.
To investigate the effect of phosphorylation on oligomerization state of LytR or LytRN, each protein was phosphorylated by acetyl phosphate as described above. Unphosphorylated and phosphorylated samples of LytRN (10 µM, 20 µM and 40 µM) were loaded onto a 10% native-PAGE. The internal temperature of the buffer during the gel electrophoresis was maintained at 4°C. The gels were stained with Coomassie blue to visualize the protein bands. We analyzed phosphorylated and unphosphorylated LytRN (80 µM) also by size exclusion chromatography TSKgel G2000SWXL (7.8 × 300mm, 5 µm, TOSOH Biosciences LLC) on HPLC.
To assess the phosphatase activity of LytS, LytRN was phosphorylated by acetyl phosphate as described above. Excess acetyl phosphate was removed by the desalting column. LytRN-P (80 µM) was incubated with 5 µM of LytS at different time intervals, in the absence or presence of 200 µM ATP, at 37°C. Samples were quenched with native-PAGE loading buffer and loaded onto a 15% native-PAGE. Native-PAGE was stained with Coomassie blue to visualize the protein bands which were quantified by NIH ImageJ.
Far UV circular dichroism (CD) spectra (200–260 nm) of LytR and LytRN were recorded using a Jasco J-180 instrument at 20°C. The buffer composition in these experiments was 20 mM Tris, 5 mM MgCl2 pH 8.0. Two spectra scans were averaged for the sample and the buffer. Later, buffer contribution was subtracted from each protein spectrum. To assess the thermal stability of LytR and LytRN, thermal melting of each protein was recorded by monitoring the change in the CD signal at 222 nm by ramping up the temperature from 20°C to 90°C at a rate of 3°C/min. Typically for these experiments, 20 µM of the respective protein was buffer exchanged into 10 mM sodium phosphate buffer at pH 7.4.
The predicted domain architectures of LytS and LytR are shown in Figure 1. LytS belongs to the family of LytS-YhcK multi-transmembrane domain bacterial receptors which carry at their intracellular C-terminal a GAF (cyclic Guanosine Monophosphate-specific phosphor-diesterases, Adenylyl cyclases and the FhlA proteins24) domain, and a kinase domain25. The amino acid analysis of LytS by InterPro (EMBL-EBI; available online at http://www.ebi.ac.uk/interpro/) predicted that this protein has five transmembrane regions (TMs), an extracellular NH2-terminus, and an intracellular GAF domain and a kinase domain. The intracellular kinase domain harbors the dimerization and histidine phosphotransfer (DHp) domain and the catalytic and ATP-binding (CA) domain.

Domain organizations of LytS (P60612) and LytR (P60609) as predicted by InterPro (EMBL-EBI) (“TM” stands for transmembrane regions; “*” denotes the phosphorylation sites, His-390 in LytS and Asp-53 in LytR; “R” stands for receiver domain; “E”, stands for the effector domain).
LytR consists of two domains, the conserved N-terminal receiver domain (LytRN) and the variable C-terminal DNA-binding domain referred to as the effector domain (LytRC). In general, the receiver domain of the RRs harbors a conserved Asp residue that undergoes a reversible phosphorylation by the cognate HK26,27. The sequence analysis of LytR reveals that the receiver domain is homologous to the receiver domains of the OmpR/NarL protein families. Based on the sequence alignments with these RRs, we determined the phosphorylation site in LytR to be Asp-53. Sequence analysis of LytR also indicates that the DNA-binding domain is homologous to that found in the novel family of non-helix-turn-helix DNA-binding domains, known as LytTR14,15. This family of proteins consists of AlgR and AgrA transcription factors15 which are involved in regulation of important virulence factors in pathogenic bacteria28. These groups of effector domains are unique in their ability to bind to DNA and account for ~2.7% of all prokaryotic RRs28.
The cloning strategy of lytS aimed to clone the cytoplasmic region of LytS spanning the amino acids 355-584, which harbors the DHp and CA domains. The cytoplasmic domain of LytS was fused at the C-terminus of a six-histidine tag (calculated molecular mass of the His-LytS is 28,359 D). The (His)6-tag aided purification of LytS to homogeneity as assessed by SDS-PAGE.
Cloning of lytR (246 amino acids) led to the expression of LytR without tags or additional amino acids (calculated molecular mass 28,221 D). Expression of LytR was good but its purification was challenging. Conventional chromatographic methods failed, and it was evident that the actual isoelectric point (pI) was different from the theoretical one (pI ~5.68). We resorted to the use of ammonium sulfate precipitation to purify the protein. The protein precipitated out at 10% ammonium sulfate and the purity was assessed to be 80%. The protein was prone to aggregation at concentrations higher than 3 mg/mL. To facilitate purification and solubility of the protein, we cloned LytR fused to the COOH-terminus of GST. GST-LytR was purified to homogeneity by affinity chromatography.
Cloning of the receiver domain of LytR, LytRN, spanning amino acids 1-134, led to the expression of a soluble protein with a molecular mass of 15,028 D without additional amino acids or tags. The protein was expressed at a higher level than LytR or GST-LytR. Moreover the protein was purified by conventional chromatographic methods based on the theoretical value of pI ~4.44. The protein was very stable and soluble at concentrations as high as 10 mg/mL.
The thermal melting points of LytR and LytRN were measured to be 55°C and 70°C, respectively. The 15 degree difference in the thermal stabilities between these two proteins is an indication that the effector domain may destabilize the N-terminal domain in the context of the full-length protein.
We monitored the level of autophosphorylation of His-LytS at different time intervals and at different ATP concentrations (Figure 2, Dataset 1). (The preliminary data were partially published as part of a poster presentation in ASBMB in April 201329.) The pseudo first-order rate constant of the autokinase activity of His-LytS was determined to be 0.030 ± 0.001 min-1. The autophosphorylation rate constant of LytS is smaller compared to the S. aureus cell wall damage sensing HK VraS (0.07 min-1)22, Enterococci faecium vancomycin resistance factor HK VanS of (0.17 min-1)30 or S. aureus essential HK WalK (0.36 min-1)31. However, it is similar to the autophosphorylation rate constants for other HKs such as E. coli nitrate sensing HK NarQ (0.014 min-1)32, and E. coli citrate sensing HK DcuS (0.043 min-1)33. The binding affinity of LytS for ATP (Ks = 7.9 ± 0.6 µM) is higher in comparison to other kinase such as, VanS (KmATP = 620 ± 42 µM)30, VraS (KmATP = 230 ± 42 µM (Belcheva et al. unpublished data)) and WalK (KmATP = 130 µM)31.
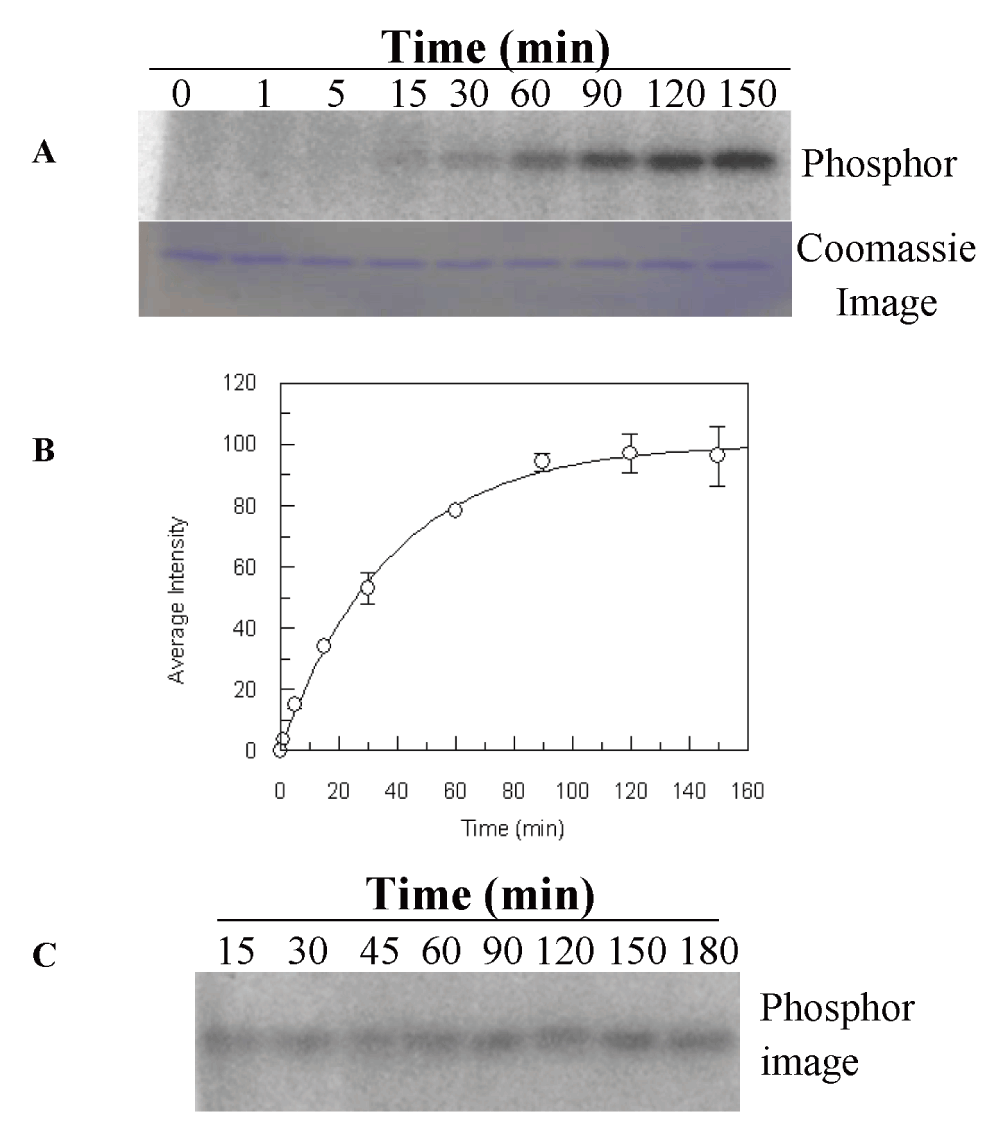
(A) His-LytS at 5 µM was incubated with [γ-32P]-ATP (100 µM) in PB at room temperature. Reaction was quenched at different time intervals and samples were analyzed by 15% SDS-PAGE. (B) Progress curve of His-LytS autophosphorylation. The quantified band intensities of phosphorylation were plotted against time. The data were fitted using Origin software to pseudo first order equation (1) to calculate the rate constant. Errors are the standard deviation from two independent trials (Dataset 1). (C) Stability of the phosphorylated His-LytS species. His-LytS at 5 µM was phosphorylated for 90 min in PB buffer (50 mM Tris, pH 7.4, 5 mM MgCl2) at room temperature. Excessive ATP was removed by desalting and stability was monitored over 3 hours at different time intervals. Samples were analyzed by 15% SDS-PAGE. All gels were exposed to phosphor-screen (GE Healthcare) overnight and imaged using a Typhoon Trio+ imager (GE Healthcare). The gel in panel (A) was also stained with Coomassie blue to view the protein bands.
We investigated phosphorylation of LytR through its cognate HK, LytS, and the small molecule phosphate donor, acetyl phosphate. Our efforts to investigate the phosphotransfer between His-LytS and LytR were hampered by the fact that the molecular masses of His-LytS and LytR were similar and this affected their separation by SDS-PAGE. To remove this obstacle, LytR was fused to GST which increased the molecular mass of LytR by 26 kD. When P32-labeled His-LytS was incubated with GST-LytR, we observed a time-dependent reduction in signal from P32-labeled His-LytS, which was associated with an increase in P32-labeling of GST-LytR (Figure 3A). The observed rate constant for the phosphotransfer process is 0.3 ± 0.1 min-1. When incubation of P32-labeled His-LytS was done in the presence of GST-LytRAsp55Asn, no reduction in signal from phosphorylated His-LytS was observed (Figure 3B). These experiments show that LytS is capable of phosphorylating LytR for as long as the phosphorylation site in LytR is available.
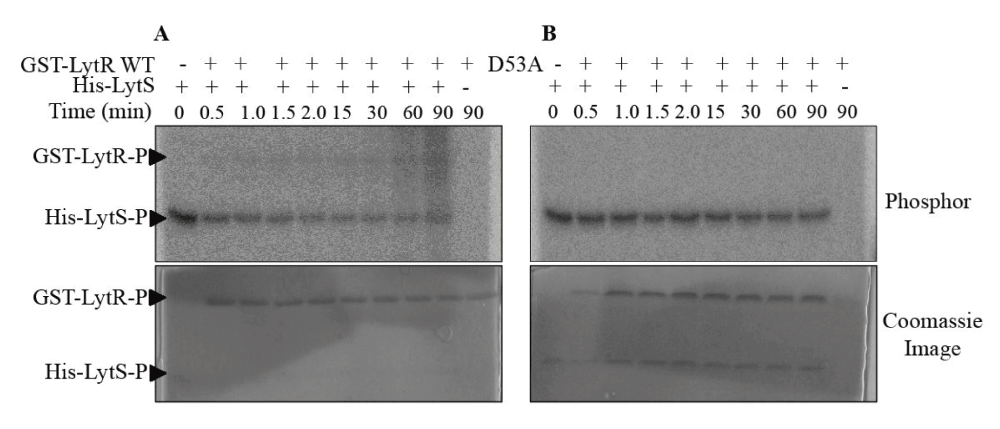
(A) Phosphorylated His-LytS at 4 µM was incubated with GST-LytR at 10 µM in PB at room temperature. The reaction was quenched at various time intervals and samples were analyzed by 15% SDS-PAGE. (B) Similar reaction as shown in (A), performed with GST-LytRAsp53Ala. The reaction was quenched at various time intervals and samples were analyzed by 15% SDS-PAGE. Gels were exposed to phosphor screen (GE Healthcare) overnight and imaged (top gels) using a Typhoon Trio+ imager (GE Healthcare) followed with Coomassie blue staining (bottom gels).
Incubation of LytR with a small molecule phosphate donor such as acetyl phosphate resulted in rapid phosphorylation of LytR (Figure 4A, Dataset 2). The observed phosphorylation rate constant for LytR was 0.6 ± 0.1 min-1. This rate is about 30-fold faster than phosphorylation of VraR by acetyl phosphate (0.022 min-1)22, or MtrA (0.014 min-1) and PrrA (0.028 min-1), 10-fold faster than DrrD (0.10 min-1), and comparable to PhoB (0.45 min-1)34. In the case of the stand-alone receiver domain, LytRN, the observed phosphorylation rate constant was 0.9 ± 0.2 min-1 (Figure 4B). The higher phosphorylation rate constant measured for the LytRN (1.5-fold compared to LytR) is another indication that the effector domain may perturb the receiver domain and very likely it does so through an interdomain interaction. Barbieri et al. recently reported a correlation between a higher phosphorylation rate constant for the receiver domains compared to the full-length RRs in cases where domains in RRs were engaged in interdomain interactions34.
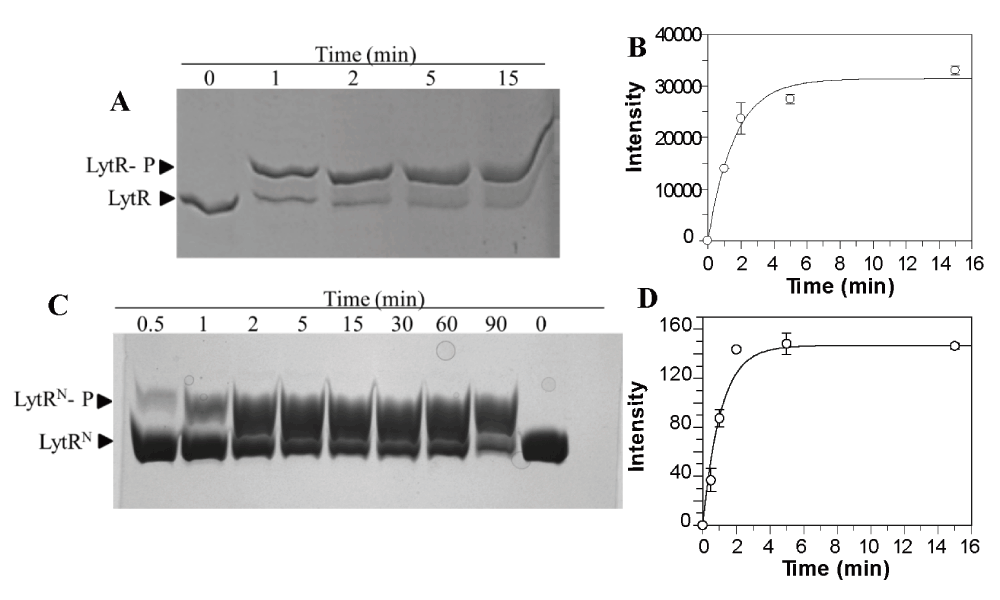
(A) LytR at 10 µM was phosphorylated by acetyl phosphate (50 mM) in PB for 1 h at 37oC. (B) Quantitative analysis of the data using NIH ImageJ (Dataset 2) The data were fitted using Origin software to pseudo first order equation (1) to calculate the rate constant. Errors are the standard deviation from two independent trials. (C) LytRN at 20 µM was phosphorylated by acetyl phosphate (50 mM) in PB buffer at 37oC and reactions were quenched at various time intervals. The phosphorylated proteins were separated from the unphosphorylated protein by 15% Phos-tag gel. The gels were stained with Coomassie blue to view the protein bands. (D) Quantitative analysis of the data using NIH ImageJ. The data were fitted using Origin software to pseudo first order equation (1) to calculate the rate constant. Errors are the standard deviation from two independent trials (Dataset 3).
Investigation of the oligomerization state of LytR was not possible through native-PAGE (Tris-Glycine, pH 8.3) as the protein was not resolved under this buffer condition. Instead, we analyzed the oligomerization state of the phosphorylated LytRN, which resolved well in native-PAGE. These experiments showed that phosphorylation of LytRN led to dimerization (Figure 5). These results can be used to predict the oligomerization state of phosphorylated LytR and it is very likely this protein dimerizes upon phosphorylation, and it does so at the receiver domain.
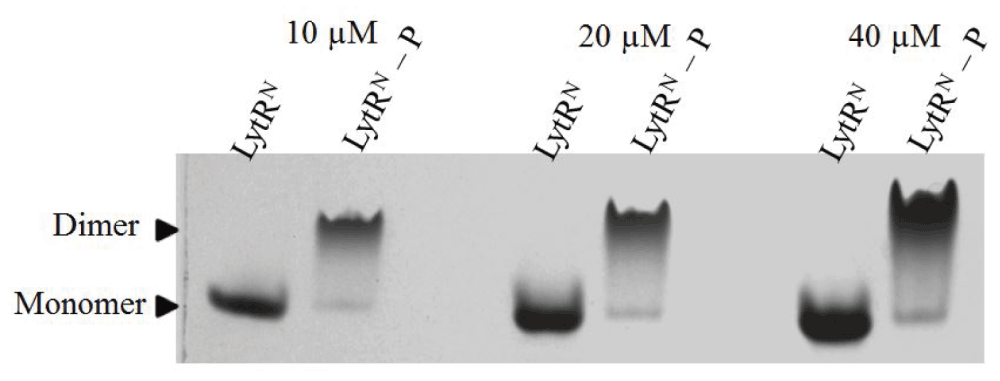
LytRN and LytRN–P at 10 µM, 20 µM and 40 µM were analyzed by 15% native-PAGE and stained with Coomassie blue.
The propensity of LytRN to dimerize upon phosphorylation was used to monitor the phosphatase activity of LytS; dephosphorylation of LytRN will lead to disintegration of the dimer which can readily be monitored in a native-PAGE system. Indeed, incubation of the phosphorylated LytRN with LytS led to conversion of the dimer to monomer species indicating that LytS has phosphatase activity (Figure 6A, Dataset 4). Interestingly, the phosphatase activity of LytS was more prominent in the presence of ATP similar to the observations made with E. coli osmosensor HK EnvZ; this phenomenon was proposed to be due to the allosteric effect that binding of ATP to CA domain had on the phosphatase activity of the DHp domain of EnvZ35.
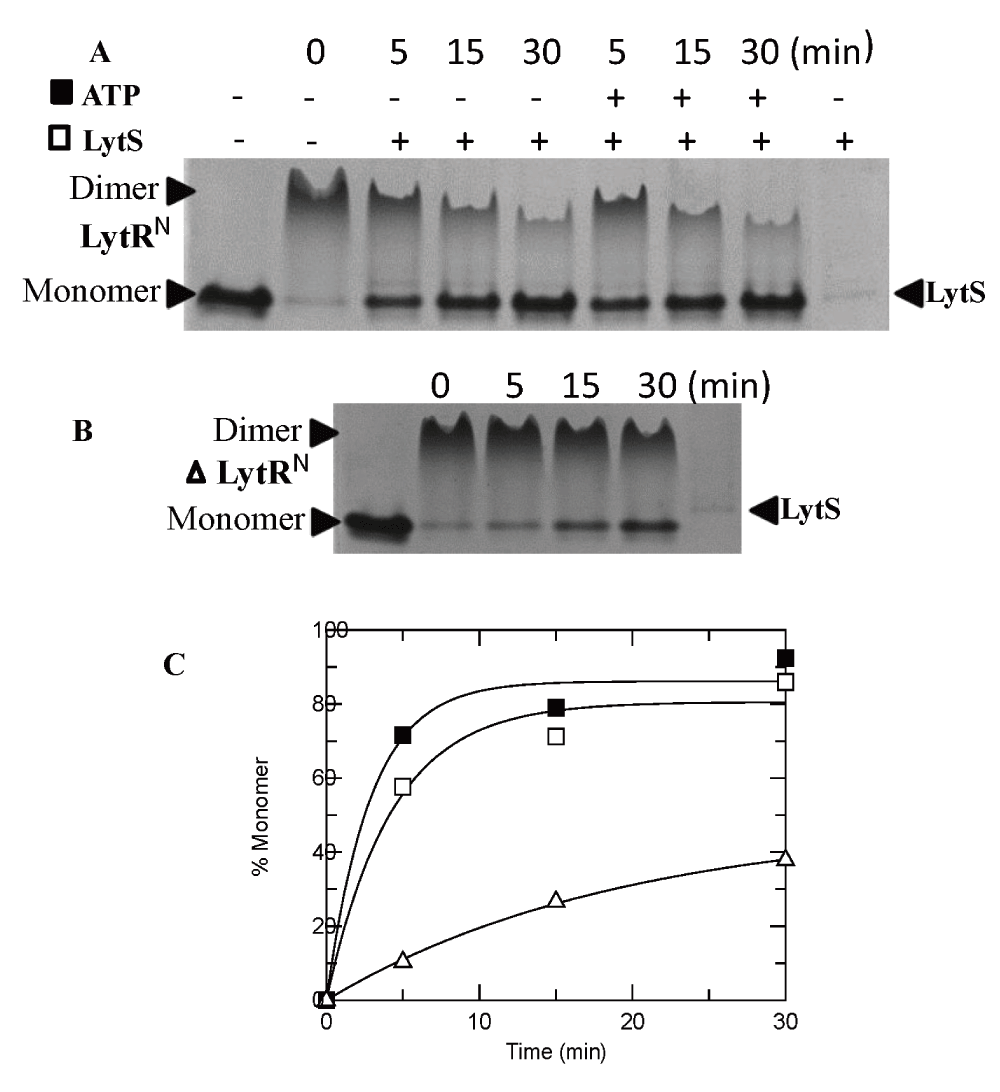
(A) LytS at 5 µM was incubated with LytRN-P at 80 µM at different time intervals, in the absence or presence of 200 µM ATP, at 37°C. (B) The stability of phosphorylated LytRN-P at 37°C. (C) Time-dependence of the phosphatase activity of LytS in the absence of ATP (empty squares) or presence of ATP (solid squares) and autophosphatase activity of LytR (empty triangles). The data points were taken from the analyses of panels A and B using NIH ImageJ (Dataset 4).
LytRN underwent slow dephosphorylation at 37°C (Figure 6B, Dataset 4). This is an indication that LytR has autophosphatase activity. However, the rate of auto-dephosphorylation of LytR is about 10% of the dephosphorylation rate by LytS (Figure 6C) hence, it may not be relevant in vivo.
LytSR is involved with sense-response to alterations of the cell electrical membrane potential due to cell membrane perturbations2,3. Its function is related to regulation of genes controlling cell apoptosis, autolysin activity and biofilm formation9,19. LytR regulates the lrgAB operon7 whereby the lrg genes products encode antiholin-like proteins that inhibit murein hydrolysis activity and cell lysis9.
To the present day, molecular details and functionality of the LytSR signal transduction pathway have only been presumed. We undertook in vitro characterization of LytS and LytR and probed their involvement in the signal transduction process. Signal transduction processes mediated by TCSs involve two reversible phosphorylation-mediated processes, autophosphorylation of the HK and transfer of its phosphoryl group to the cognate RR. Quite often the role of HK is to control the phosphorylation level of RRs, which in turn regulates the transcriptional activity of RR. It does so by possessing phosphatase activity toward RR, in addition to the kinase activity13. The phosphorylation level of RRs, however, can also be regulated through phosphorylation by small molecule phosphate donors such as acetyl phosphate and the autophosphatase activity of RRs26.
Our study demonstrates that the cytoplasmic domain of LytS has autokinase activity (k = 0.030 ± 0.001 min-1). The LytS phosphorylation rate constant is comparable to other HKs. The unusually low dissociation constant measured for ATP with LytS in comparison to other RRs, such as VraR, WalK, or VanS, suggests that the autophosphorylation efficiency for LytS is high and the kinase is well positioned to participate directly in the signalling process induced by changes in the cytoplasmic membrane electrical potential. The fast phosphotransfer process that we observed between LytS and LytR (0.3 min-1) suggests that any alteration in the cell membrane electrical potential sensed by LytS is efficiently transduced intracellularly.
It is well established that most RRs are also equipped with the ability to catalyze their own phosphorylation, independently of their cognate kinases, using endogenous low molecular weight phosphoryl group donors36. In fact, phosphorylation of RRs by low molecular weight phosphoryl group donors such as acetyl phosphate, carbamyl phosphate or phosphoramide appears to be more common than phosphorylation by non-cognate HKs37. Intracellular concentration of acetyl phosphate ranges from 1 mM to 3 mM in vivo, suggesting that this phosphate group donor is available in the cell in similar quantities as ATP34,36,37. Herein, we show that LytR undergoes rapid and quantitative phosphorylation by acetyl phosphate (k = 0.6 min-1). Further, phosphorylation of the receiver domain by acetyl phosphate leads to dimerization of this domain demonstrating that phosphorylation-induced activation of LytR involves formation of dimers at the receiver domain. The differences in the thermal denaturation and phosphorylation rates by acetyl phosphate of LyR and LytRN provide evidence that the effector domain has a destabilizing effect on the receiver domain, which could plausibly be due to an interdomain interaction.
The rapid phosphorylation of LytR by acetyl phosphate observed in our study (about 2-fold faster than phosphorylation by LytS) strongly suggests that this pathway is important in vivo. Interestingly, in vivo studies have demonstrated presence of two overlapping regulatory networks in regulation of the lrgAB operon1,20. One regulatory network responds to excess glucose metabolism and the other responds to changes in the cell membrane potential2. Induction of lrgAB in response to glucose metabolism was shown in vivo to rely on LytR2, however, metabolism of excess glucose does not lead to changes in the cytoplasmic membrane electrical potential1, hence it is less likely that signaling occurs through LytS in this case. The fast kinetics of LytR phosphorylation by acetyl phosphate makes this pathway an efficient signaling and regulatory mechanism of lrgAB in response to the glucose metabolism. Moreover, the phosphatase activity of LytS toward phosphorylated-LytR, which otherwise is stable during the cell division time, provides the regulatory means to shut-down this pathway when the glucose level in the medium goes down.
In summary, our study shows that LytSR can participate in two signal transduction pathways through two phosphorylation processes: phosphorylation of LytR by LytS and phosphorylation of LytR by acetyl phosphate (Figure 7). Further, our findings provide the molecular mechanism for the in vivo observation that regulation of lrgAB operon is LytR dependent, either in response to excess of glucose metabolism or perturbation of cell membrane electrical potential.
A recent report by Lehman et al. showed that phosphorylation of LytR by acetyl phosphate is relevant in vivo and it is important for regulation of lrgAB operon under high level of glucose38.
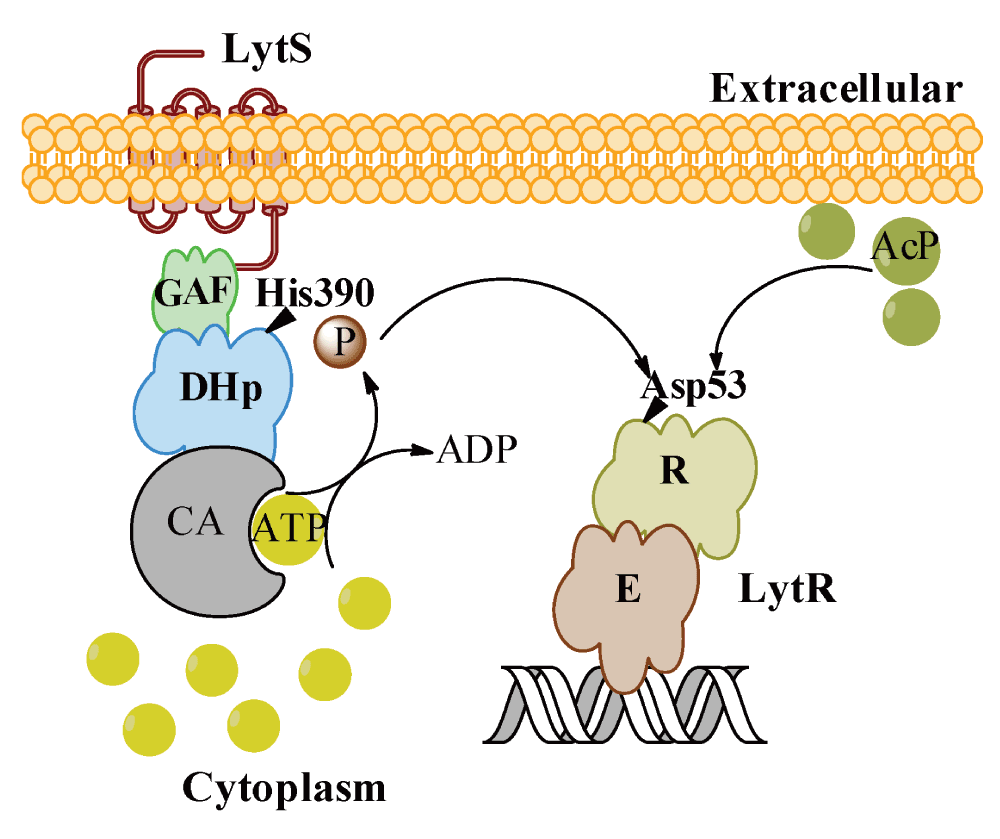
Our data support two pathways of activation of LytR: through LytS as a result of perturbation of the membrane electric potential, and through acetyl phosphate as a result of accumulation of acetate during metabolism of excess glucose.
Figshare: Raw data for the role of acetyl phosphate in bypassing the cell membrane electrical potential sensor LytS doi: 10.6084/m9.figshare.133984339
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)