Keywords
photosynthesis, light harvesting, click chemistry, Rhodobacter sphaeroides, Roseobacter denitrificnas, Phaeosprillum rubrum, core complex
photosynthesis, light harvesting, click chemistry, Rhodobacter sphaeroides, Roseobacter denitrificnas, Phaeosprillum rubrum, core complex
The light harvesting apparatus of photosynthetic bacteria, and indeed of all photosynthetic organisms, is organized in a large array of complex structure. This array is designed to absorb light energy and efficiently funnel it to available reaction centers1. In most purple bacteria the light collection array is comprised of two proteins: the reaction center containing core complex (CC), and the peripheral light harvesting complex (LH2). In Figure 1A an AFM image of a fragment of photosynthetic membrane from Rhodospirillum photometricum is shown, immediately apparent is the extensive array of proteins. The visible proteins are of two types: smaller rings composed of LH2, a molecular model of which is shown in panel B, and larger rings composed of CC, a molecular model of which is shown in panel C. Analysis of the organization of these proteins2 has shown that this organization can be described as a mixture of hexagonal packed arrays of the nonameric LH2, and a more randomly organized LH2 CC mixture.
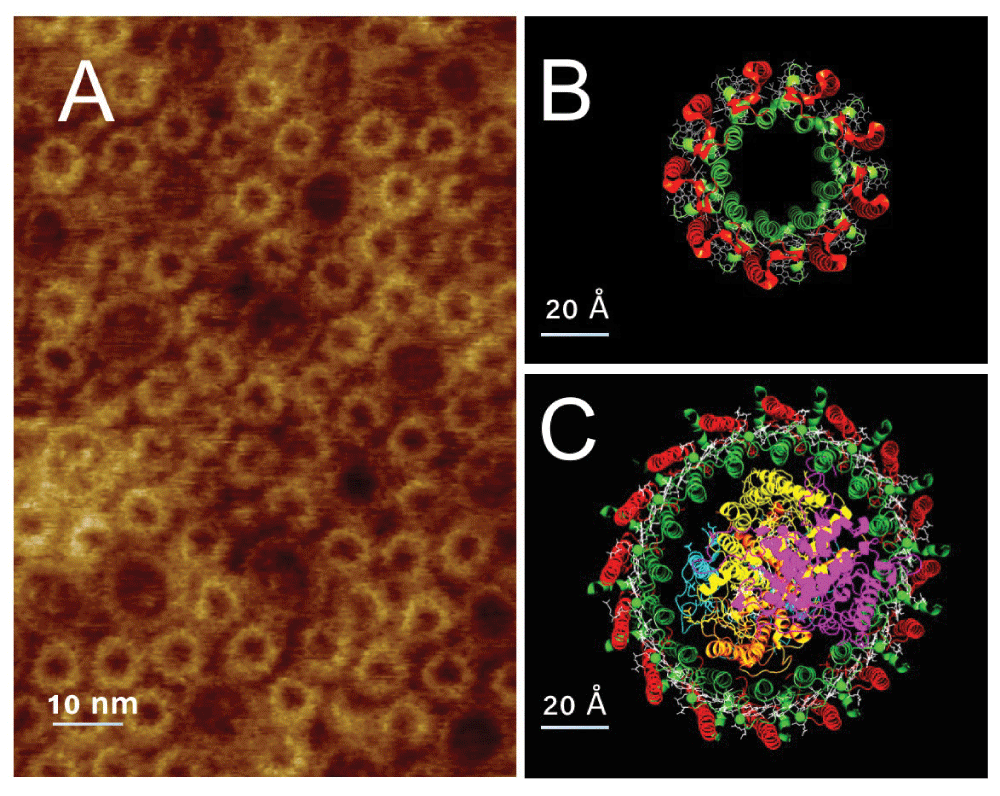
A, AFM image of a fragment of photosynthetic membrane from Rhodospirillum photometricum showing the organization of LH2 (small rings) and CC (larger rings) in the membrane. B, Molecular model of LH2 viewed perpendicular to the membrane surface, the α polypeptides in green and β in red. C, Similar view of a molecular model of the CC, again the α subunits are in green and the β in red, the reaction center C,L,M and H subunits are in magenta, orange, yellow and cyan respectively.
The individual protein rings shown in Figure 1B & 1C are each oligomeric. The LH2 ring (Figure 1B) is made of a circular array, of usually 9, subunits each composed of two polypeptides α and β, shown in green and red respectively, which bind four pigment molecules, 3 bacteriochlorophyll (BChl) a and one carotenoid molecule3,4. In the case of Phaeospirillum (Phsp.) molischianum the ring is composed of 8 subunits5. The CC is slightly more complex, and also variable, in architecture. In the center is a reaction center component, typically made of the subunits H, M and L with a bound tetraheme cytochrome subunit C, these are shown in Figure 1C colored cyan, yellow, orange and magenta respectively. This is surrounded by the light harvesting complex oligomer of 16 α and β subunits. These subunits are similar to those of LH2 but only bind 2 BChl a molecules6. There is considerable inter-species variability in the precise organization of the CC, with variants having less than 16 subunits, and additional subunits that do not bind pigment or complexes forming S-shaped dimers7.
In order to better understand energy flow within the light harvesting array8 it would be useful to study isolated parts of the array, larger than a single complex but smaller than the whole array, and with defined architecture.
This seemingly simple task has proved surprisingly complex. The main difficulties are the large size of the array in situ, the oligomeric structure of the pigment-protein complexes and the need to use detergents as membrane proteins are insoluble in standard biochemical buffers. Initial experiments9,10 with chemical cross-linking in membranes gave very complex mixtures of intra-molecularly linked proteins, inter-molecularly linked homodimers and heterodimers with altered purification properties rendering the objective of reasonably homogeneous samples of defined architecture unattainable.
Here we develop a protocol that allows us to form small relatively homogeneous oligomers (dimers and trimers) in a controlled manner. We show that the yields for the different steps are reasonable and that the end product is as expected.
The various pigment protein complexes were purified by standard methods as described previously11. Briefly photosynthetic membranes were solubilized with dodecyl-maltoside and pigment protein complexes isolated by sucrose density gradient centrifugation followed by anion exchange chromatography on a resource-Q column and gel filtration on a superose-6 column. The purity of the various complexes was evaluated by absorption spectroscopy (Shimadzu UV1800 spectrophotometer), measuring the ratio of the absorption peaks at 280 nm and 370 nm. This ratio varies somewhat depending on the source, but absorption ratios of 0.3 and 0.65 are typical for purified LH2 and CC respectively. Purified LH2 and CC proteins from Phsp. molischianum, Rhodobacter (Rb.) sphaeroides and Roseobacter (Rsb.) denitrificans were prepared in 20 mM sodium phosphate buffer (pH 7.2) containing 0.05% dodecyl maltoside. For each purified complex extinction coefficients were calculated from a knowledge of the stoichiometry, thanks to the known structures of the complexes, and BChl a extraction from purified complexes as described previously12. Briefly, the UV-visible absorption spectrum was measured, the BChla concentration of the sample was determined from the absorption of an acetone methanol (7:2) extract using the extinction coefficient of ϵ770nm 76 mM–1cm–113.
The stoichiometry of the complex was calculated as 4 BChl per reaction center plus 2 BChl per LH1 type αβ pair for core complexes and 3 BChl per LH2 type αβ pair for LH2 complexes. The complex extinction coefficient was then calculated as ϵ(cm–1mM–1) = Absorbance ͙ stoichiometry/(1cm͙ [BChl]), see Dataset 1.
Succinimidyl esters, (5/6-carboxyfluorescein succinimidyl ester and succinimidyl-2-(biotinamido)ethyl-1,3- dithiopropionate), were purchased from Thermo Fisher Scientific (Waltham, USA) and dissolved in dry DMSO from Acros (Geel, Belgium) prior to use. Primary amine labeling was carried out at 4°C for 1 hour in 20 mM Na Phosphate buffer pH 7.2 containing 0.05% dodecyl maltoside.
Maleimides, dibenzylcyclooctyne-PEG4-maleimide and azido-PEG3-maleimide, were purchased from Jena Bioscience and dissolved in dry DMSO prior use. Sulfhydryl labeling was carried out at 25°C for 2 hours in 20 mM Na Phosphate buffer pH 7.2 containing 0.05% dodecyl maltoside. Coupling by copper-free click chemistry was performed in the same buffer for 10 hours at 4°C.
After reaction with 5/6-carboxyfluoresceine succinimidyl ester and the maleimides the labeled protein was separated from unreacted label using spin columns (Micro Biospin TM6 columns, Bio-Rad (Hercules,USA)), according to the manufacturer’s instructions.
Reaction products after coupling were analyzed by HPLC. 20–40 μl samples were injected and separated on a Agilent technologies (Santa Clara, CA) 1260 infinity chromatography system equiped with a Biosep-SEC-s4000 analytical column (300mmx4.60mm) eluted with 20 mM Na Phosphate buffer pH 7.2 containing 0.05% dodecyl maltoside at a flow rate of 0.5 ml/min and followed by absorption at 280 nm. Absorption spectra of peaks were obtained from the integrated spectral detector (Agilent technologies G1315D diode array detector).
Molecular models of proteins were prepared based on homology to proteins of known structure. In the case of LH2 the structures of the proteins from Rhodopseudomonas (Rps.) acidophila14, and Phsp. molischianum5 were used as templates. For core complexes the recent structure of the protein from Allochromatium (Ac.) tepidum15 was used as a template.
Model structures were visualized and diagrams prepared with Pymol molecular graphics program, version 1.816.
The general approach that we used is shown in Figure 2. In the first step the most reactive lysines in the complex are reacted with a succinimidyl-ester at a very low degree of labeling to ensure on average less than 1 reacted lysine per complex. This low degree of labeling is essential to ensure that during cross-linking the number of higher order oligomers formed is minimal. The choice of an amine directed reagent was governed by the known structure of the complexes where, thanks to the positive inside rule17, a number of lysines predicted to be reactive are routinely found in the exposed cytoplasmic parts of the light harvesting ring. Indeed examination of the structures of LH2 and CC show that there is a ring of exposed lysines close to the cytoplasmic surface of the membrane.
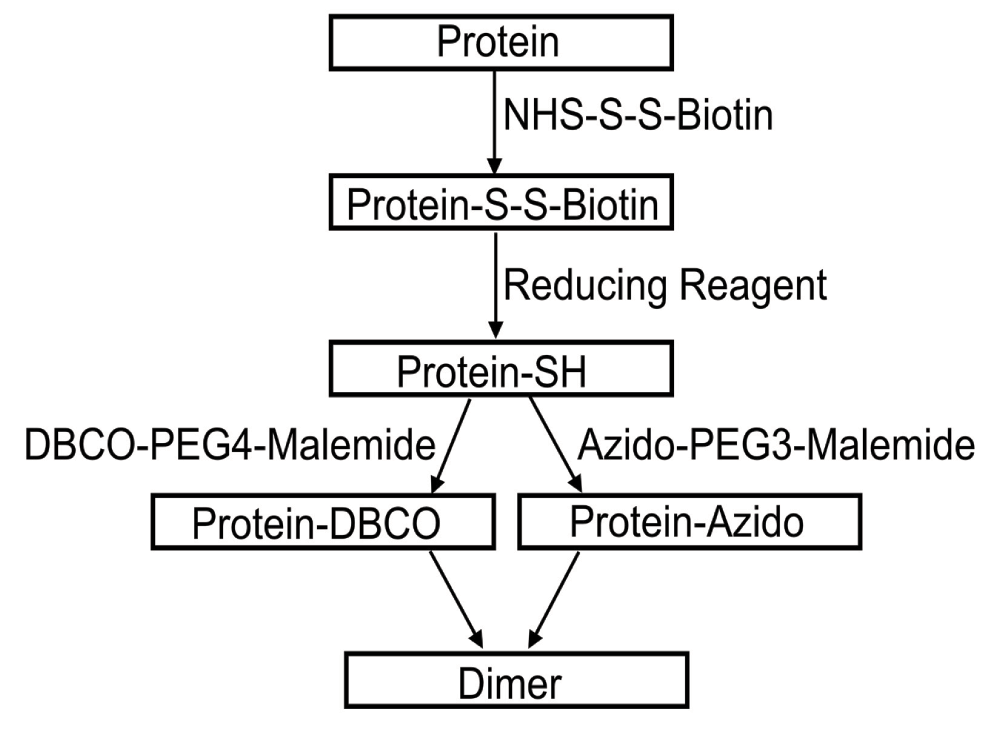
Complexes appropriate for copper-free click chemistry, labeled with DBCO- or azido- groups, were prepared in a two step procedure that allows affinity purification of a biotinylated intermediate.
Singly labeled proteins prepared by partial reaction of lysines were then purified, thanks to the use of a biotin containing reagent, by affinity on streptavidin beads, and eluted by reduction of the dithiol linkage in the reagent. The resulting proteins contain a unique reactive thiol, the proteins used do not have exposed cysteines. So in the next step of the protocol the thiols are reacted with maleimides appropriate for click chemistry. Allowing the formation of self-non reactive singly labeled proteins that can react with each other, an azido-labeled protein with an alkyne-labeled one.
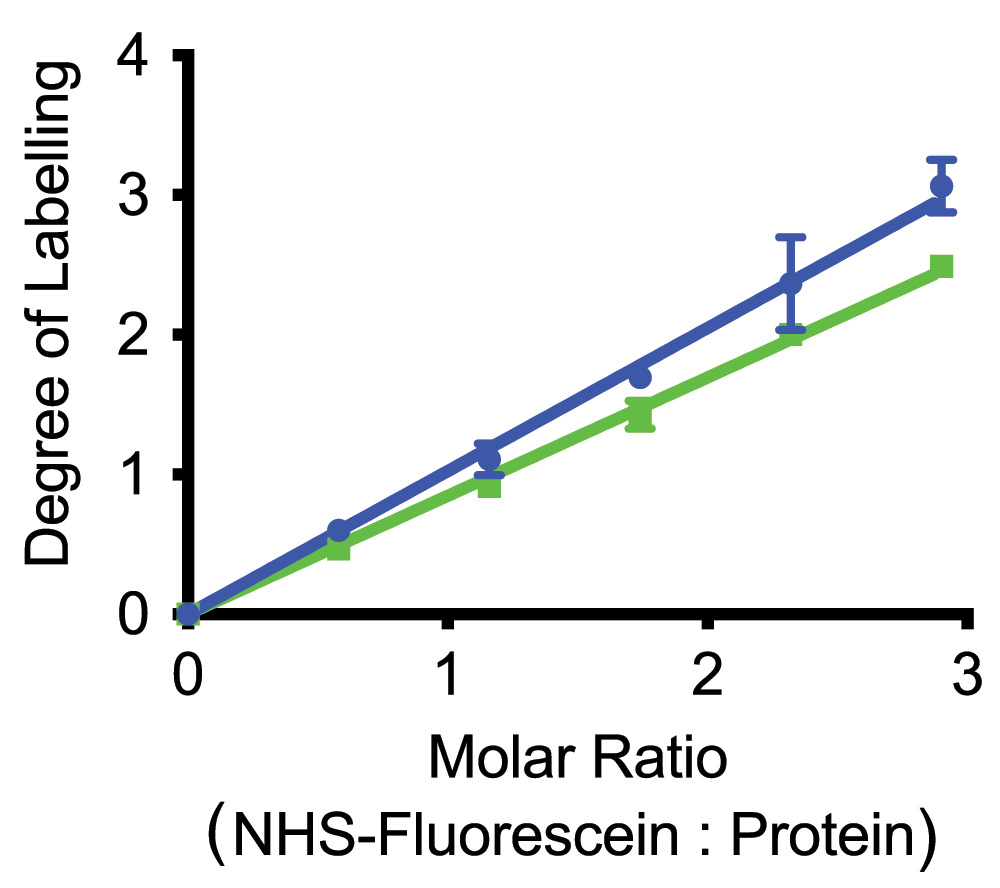
Curves are shown for two different purified complexes: LH2 of Phsp. molischianum (green), and CC of Phsp. molischianum (blue) (Dataset 2).
Critical for the general protocol outlined above (Figure 2) is the possibility of obtaining protein with a controlled low level of labeling on lysine residues. To assess this and verify that even under these somewhat non standard conditions we could obtain reproducible labeling we used carboxyfluoresceine succinimidyl ester to follow the degree of labeling. In Figure 3 we show the degree of labeling observed by absorption spectroscopy using the most red Bchl a absorption band, with the extinction coefficients calculated as described above, and the fluorescine absorption at 494 nm, and the published extinction coefficient of 70 mM–1cm–118. This graph shows that the degree of labeling was linear in the amount of succinimidyl-ester added without any noticeable threshold for labeling. The slopes of the relationship varied between proteins, presumably reflecting differences in the reactivity of the targeted lysines. In Figure 3 we show curves for CC (blue) and LH2 (green) of Phsp. molischianum, the slopes of the labeling relationship for all the different complexes we examined vary over a relatively narrow range of 0.98 to 0.81.
As the first step was designed to give a very low level of labeling, limited by the amount of succinimidyl ester, it was necessary to separate the labeled protein from unlabeled protein. To achieve this efficiently we chose to use succinimidyl-2-(biotinamido)ethyl-1,3- dithiopropionate, this reagent allows us to strongly and specifically bind labeled protein to streptavidin beads, and elute the protein either with biotin or by reducing the disulfide bond.
Tests showed that, as expected, the binding was specific and typical yields with unlabeled protein, containing no bound biotin groups, was less than about 0.1% (the estimated detection limit). In contrast estimated yields of biotin containing proteins were greater than 95%.
We chose to use disulfide reduction to elute the proteins from the streptavidin beads, this approach gave better yields for the reacted proteins. Two different reducing agents were tested: tris(2-carboxyethyl)phosphine (TCEP) and dithiothreitol (DTT). Both proved able to release bound reacted proteins. However some proteins, notably several core complexes, proved rather sensitive to TCEP reduction losing their colored cofactors. We therefore decided to use DTT as the reductant, adding DTT to a final concentration of 50mM and allowing reduction to proceed for 4 hours at 25°C. The eluted protein was separated from excess reductant by gel filtration on a spincolumn.
The selectivity and specificity of this purification method ensures that the majority of the eluted protein, even at very low labeling levels will have one or more reacted lysines. For example, if a degree of labeling of 10% is targeted for a nonameric protein the probability of labeling on of the 9 equivalent most reactive lysines is 1.1% from this, assuming independent labeling of the different groups, we expect the reaction mixture to contain 90.4% unlabeled protein, 9.1% singly labeled protein and 0.5% multiply labeled protein. After purification, given the selectivity and specificity, the mixture is expected to contain less than 1% unlabeled protein and less than 5% doubly labeled protein and 94.6% singly labeled protein.
Initial attempts to cross-link complexes using copper catalyzed click chemistry indicated that the colored cofactors were rather sensitive to monovalent Cu and lost their color under typical reaction conditions. Again this was particularly noticeable for CC and the B800 absorption band of LH2 complexes. In view of this we decided to use a copper-free click chemistry protocol for cross linking based on the strained cyclo-octyne ring19. Aliquots of the proteins to cross-link were reacted with a 50 fold excess of dibenzylcyclooctyne-PEG4-maleimide or azido-PEG3-maleimide at room temperature for 1.5 hours. Unreacted reagent was removed by passage of the reaction mix over a spin column.
Immediately after preparation of the azide and cyclooctane derivatives they were mixed in a 1:1 molar ratio and allowed to react at 4°C over night. After the reaction was complete the sample was analyzed by HPLC.
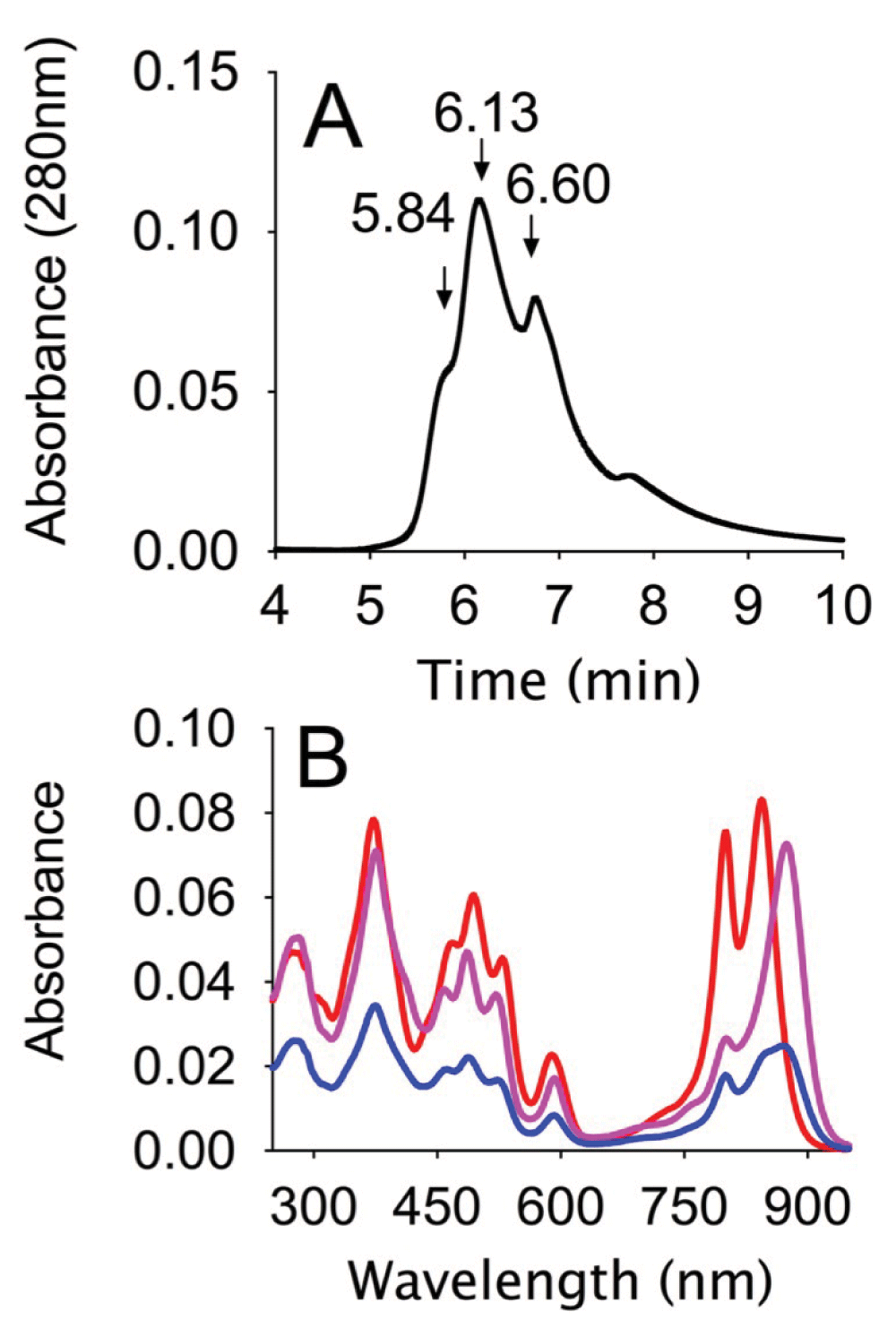
A, HPLC trace showing absorption at 280 nm of the reaction mixture separated on an analytical Biosep-SEC-s4000, in 20 mM Na phosphate buffer pH 7.2, containing 0.05% Dodecyl maltoside, at a flow rate of 0.5ml/min (Dataset 3). B, Absorption spectra of the major peaks in the HPLC trace: 5.84 min (blue), 6.13 min (magenta), 6.60 min (red) (Dataset 4).
Reaction products were analyzed by HPLC on a size exclusion column. Typically, as shown in Figure 4A, several peaks could be observed, the first peak eluting at 5.84 min is specific to samples in which the alkyne and azide were present and corresponds to the cross-linked product. This is followed by peaks, at 6.13 and 6.6 minutes, corresponding to the unreacted CC and LH2 proteins respectively. Several smaller peaks can be observed in the later part of the chromatogram.
The identity of the different peaks was confirmed by absorption spectroscopy as can be seen in Figure 4B. The second and third peaks show the expected absorption of CC and LH2 respectively, while the first peak shows an absorption peak typical of a mixture of LH2 and CC. The absorption spectra of the first peak, when analyzed using the extinction coefficients determined for the individual pigment protein complexes gives a CC:LH2 ratio of 1.5, this would suggest either some contamination due to poor separation from CC and/or some higher order oligomers containing more than one CC attached to an LH2.
The presence of relatively large amounts of monomeric proteins and several peaks of low molecular components with UV absorption was neither expected nor desired. The most likely explanation of this is poor separation of labeled protein from unreacted or click chemistry reagents coupled with poor yield in the reaction between the maleimides and the thiol-containing protein. Unfortunately we have not as yet been successful in addressing this yield issue. Nevertheless, the protocol we have developed allows to form and separate heterodimers of several different proteins in a controlled manner.
While the polypeptides of certain LH2 complexes, for example those of Rb. sphaeroides or Rsb. denitrificans only contain lysine residues in the N-terminal, cytoplasmic, part of the sequence others such as those from Rps. acidophila or Phsp. molischianum have lysines in both the N and C terminal portions. Equally in core complexes there are lysines in the N terminal regions of the core antennae but also, depending on the species, various other potentially reactive lysines in the reaction center subunits and occasionally in the C-terminal region. To better understand the structure of the dimers formed, and assess their biological relevance, it would be useful to determine which lysines are linked together by cross-linking. Unfortunately our attempts to locate the labeling positions the labeling positions in the LH2 and core complexes from Phsp. molischianum, by MALDI-MS following digestion with various proteases (trypsin, chymotrypsin of V8 protease) were unsuccessful. Mass spectometry measurements were made on a Bruker Microflex II in positive reflectron mode with automatic sampling and peak identification followed by manual verification. The inability to determine the site of labeling in core complexes at low degrees of labeling, suggests perhaps that labeling is on the more numerous light harvesting polypeptides, since no difference was observed in the reaction center polypeptides detected before and after reaction even with degrees of labeling above 1. This however remains circumstantial and highly tentative.
The general protocol we propose is able to produce purified dimeric, and possibly trimeric, complexes from the proteins of the purple bacterial photosynthetic apparatus. The approach based on reaction at a very low degree of labeling to ensure essentially mono-derivatized oligomers, despite the large number of equivalent reactive groups, followed by high selectivity and specificity purification of the activated proteins and copper-free click chemistry is able to overcome several of the problems we have previously encountered in trying to obtain such complexes. Notably the formation of highly cross-linked products is not observed, such multimeric products would be expected to elute earlier from the gel filtration column (4 min). The destruction of pigments inherent to copper (I) based cyclo-addition was also avoided.
Nevertheless the approach is not without problems and two are particularly flagrant. First the yield in the final step is much less than expected, and appears to be rather variable. Second we have also been unsuccessful in determining the site of labeling.
The low yield in the final step could derive from several different causes. First, the presence of contaminant DBCO and Azide after purification of the labeled protein, could lead to side reactions inactivating the labeled proteins. Second, the reactivity of the Cu free reagents could lead to deactivation/reaction before the mixing of the two proteins. Third, the maleimide labeling could be less efficient than expected. We suspect at least the first two possibilities are partly responsible, but have been unable to resolve the issue.
The difficulty in determining the site of labeling is the consequence of 2 different factors. On the one hand membrane proteins are often rather hard to study by mass spectrometry, and this results in low yields and coverage with proteomic approaches and difficulties in obtaining total mass due to the presence of detergents.
The site of labeling of the different proteins does however need to be confirmed to build models of such complexes and understand their behavior. This absence of knowledge is less important for certain complexes, for example as mentioned above the LH2 of Rsb. denitrificans has only two lysines in the N-terminal portion of the protein, α5 and β720, thus we can be fairly certain labeling is close to the cytoplasmic side of the protein. Unfortunately for the majority of complexes such simple deduction is not possible. However for the core complex we have some circumstantial indications that labeling is of the more abundant light harvesting polypeptides as we do not observe changes in the mass spectrum of the reaction center components at a degree of labeling of 1.0.
We have developed a novel protocol based on copper-free click chemistry that allows the formation and purification of specific dimers between highly oligomeric proteins. This protocol can be used to prepare biologically relevant dimers of certain LH2s with very asymmetrical lysine distribution, for example the LH2 of Roseobacter denitrificans or Rhodobacter sphaeroides. Such dimers will be of considerable interest for studying energy migration in light-harvesting arrays.
F1000Research: Dataset 1. Extinction coefficients for purified complexes, 10.5256/f1000research.8676.d12134921
F1000Research: Dataset 2. Degree of labeling for different complexes, 10.5256/f1000research.8676.d12135022
F1000Research: Dataset 3. HPLC trace after cross-linking, 10.5256/f1000research.8676.d12135123
F1000Research: Dataset 4. Spectra of peaks in HPLC profile, 10.5256/f1000research.8676.d12135224
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)