Keywords
methylation, bioconductor, workflow, array
This article is included in the Bioconductor gateway.
methylation, bioconductor, workflow, array
DNA methylation, the addition of a methyl group to a CG dinucleotide of the DNA, is the most extensively studied epigenetic mark due to its role in both development and disease (Bird, 2002; Laird, 2003). Although DNA methylation can be measured in several ways, the epigenetics community has enthusiastically embraced the Illumina HumanMethylation450 (450k) array (Bibikova et al., 2011) as a cost-effective way to assay methylation across the human genome. More recently, Illumina has increased the genomic coverage of the platform to >850,000 sites with the release of their MethylationEPIC (850k) array. As methylation arrays are likely to remain popular for measuring methylation for the foreseeable future, it is necessary to provide robust workflows for methylation array analysis.
Measurement of DNA methylation by Infinium technology (Infinium I) was first employed by Illumina on the HumanMethylation27 (27k) array (Bibikova et al., 2009), which measured methylation at approximately 27,000 CpGs, primarily in gene promoters. Like bisulfite sequencing, the Infinium assay detects methylation status at single base resolution. However, due to its relatively limited coverage the array platform was not truly considered “genome-wide” until the arrival of the 450k array. The 450k array increased the genomic coverage of the platform to over 450,000 gene-centric sites by combining the original Infinium I assay with the novel Infinium II probes. Both assay types employ 50bp probes that query a [C/T] polymorphism created by bisulfite conversion of unmethylated cytosines in the genome, however, the Infinium I and II assays differ in the number of beads required to detect methylation at a single locus. Infinium I uses two bead types per CpG, one for each of the methylated and unmethylated states (Figure 1a). In contrast, the Infinium II design uses one bead type and the methylated state is determined at the single base extension step after hybridization (Figure 1b). The 850k array also uses a combination of the Infinium I and II assays but achieves additional coverage by increasing the size of each array; a 450k slide contains 12 arrays whilst the 850k has only 8.
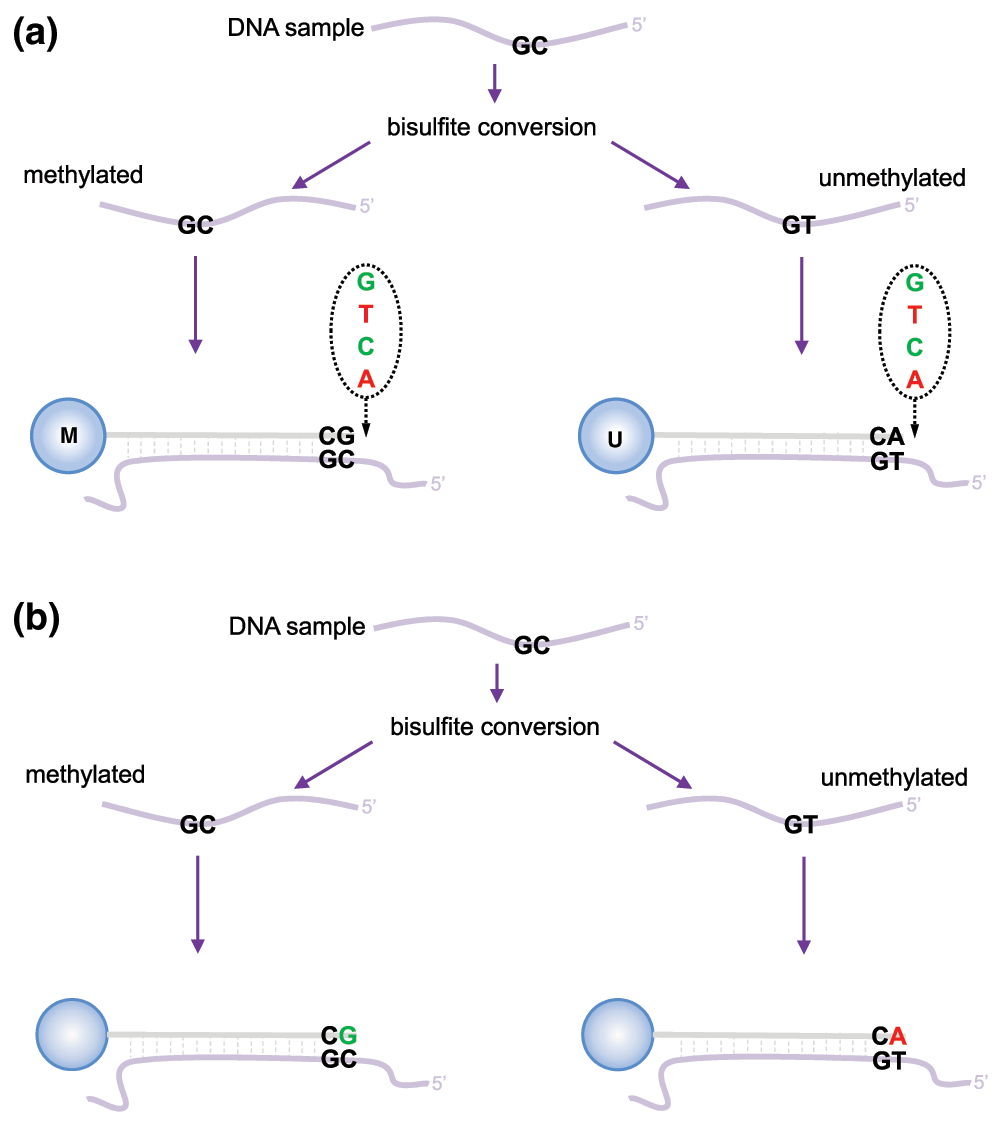
(a) Infinium I assay. Each individual CpG is interrogated using two bead types: methylated (M) and unmethylated (U). Both bead types will incorporate the same labeled nucleotide for the same target CpG, thereby producing the same color fluorescence. The nucleotide that is added is determined by the base downstream of the ‘C’ of the target CpG. The proportion of methylation can be calculated by comparing the intensities from the two different probes in the same color. (b) Infinium II assay. Each target CpG is interrogated using a single bead type. Methylation state is detected by single base extension at the position of the ‘C’ of the target CpG, which always results in the addition of a labeled ‘G’ or ‘A’ nucleotide, complementary to either the ‘methylated’ C or ‘unmethylated’ T, respectively. Each locus is detected in two colors, and methylation status is determined by comparing the two colors from the one position.
Regardless of the Illumina array version, for each CpG, there are two measurements: a methylated intensity (denoted by M) and an unmethylated intensity (denoted by U). These intensity values can be used to determine the proportion of methylation at each CpG locus. Methylation levels are commonly reported as either beta values (β = M/(M+U+α)) or M-values (Mvalue = log2(M/U)). Beta values and M-values are related through a logit transformation. Beta values are generally preferable for describing the level of methylation at a locus or for graphical presentation because percentage methylation is easily interpretable. However, due to their distributional properties, M-values are more appropriate for statistical testing (Du et al., 2010).
In this workflow, we will provide examples of the steps involved in analysing methylation array data using R (R Core Team, 2014) and Bioconductor (Huber et al., 2015), including: quality control, filtering, normalization, data exploration and probe-wise differential methylation analysis. We will also cover other approaches such as differential methylation analysis of regions, differential variability analysis, gene ontology analysis and estimating cell type composition. Finally, we will provide some examples of useful ways to visualise methylation array data.
To demonstrate the various aspects of analysing methylation data, we will be using a small, publicly available 450k methylation dataset (Zhang et al., 2013). The dataset contains 10 samples in total; there are 4 different sorted T-cell types (naive, rTreg, act_naive, act_rTreg), collected from 3 different individuals (M28, M29, M30). For details describing sample collection and preparation, see Zhang et al. (2013). An additional birth sample (individual VICS-72098-18-B) is included from another study (Cruickshank et al., 2013) to illustrate approaches for identifying and excluding poor quality samples.
targets[,c("Sample_Name","Sample_Source", "Sample_Group")]
## Sample_Name Sample_Source Sample_Group
## 1 1 M28 naive
## 2 2 M28 rTreg
## 3 3 M28 act_naive
## 4 4 M29 naive
## 5 5 M29 act_naive
## 6 6 M29 act_rTreg
## 7 7 M30 naive
## 8 8 M30 rTreg
## 9 9 M30 act_naive
## 10 10 M30 act_rTreg
## 11 11 VICS-72098-18-B birthThere are several R Bioconductor packages available that have been developed for analysing methylation array data, including minfi (Aryee et al., 2014), missMethyl (Phipson et al., 2016), wateRmelon (Pidsley et al., 2013), methylumi (Davis et al., 2015), ChAMP (Morris et al., 2014) and charm (Aryee et al., 2011). Some of the packages, such as minfi and methylumi include a framework for reading in the raw data from IDAT files and various specialised objects for storing and manipulating the data throughout the course of an analysis. Other packages provide specialised analysis methods for normalisation and statistical testing that rely on either minfi or methylumi objects. It is possible to convert between minfi and methylumi data types, however, this is not always trivial. Thus, it is advisable to consider the methods that you are interested in using and the data types that are most appropriate before you begin your analysis. Another popular method for analysing methylation array data is limma (Ritchie et al., 2015), which was originally developed for gene expression microarray analysis. As limma operates on a matrix of values, it is easily applied to any data that can be converted to a matrix in R.
We will begin with an example of a probe-wise differential methylation analysis using minfi and limma. By probe-wise analysis we mean each individual CpG probe will be tested for differential methylation for the comparisons of interest and p-values and moderated t-statistics will be generated for each CpG probe.
It is useful to begin an analysis in R by loading all the package libraries that are likely to be required.
# load packages required for analysis library(limma) library(minfi) library(IlluminaHumanMethylation450kanno.ilmn12.hg19) library(IlluminaHumanMethylation450kmanifest) library(RColorBrewer) library(missMethyl) library(matrixStats) library(minfiData) library(Gviz) library(DMRcate) library(stringr)
The minfi package provides the Illumina manifest as an R object which can easily be loaded into the environment. The manifest contains all of the annotation information for each of the CpG probes on the 450k array. This is useful for determining where any differentially methylated probes are located in a genomic context.
# get the 450k annotation data ann450k = getAnnotation(IlluminaHumanMethylation450kanno.ilmn12.hg19) head(ann450k)
## DataFrame with 6 rows and 33 columns
## chr pos strand Name AddressA
## <character> <integer> <character> <character> <character>
## cg00050873 chrY 9363356 - cg00050873 32735311
## cg00212031 chrY 21239348 - cg00212031 29674443
## cg00213748 chrY 8148233 - cg00213748 30703409
## cg00214611 chrY 15815688 - cg00214611 69792329
## cg00455876 chrY 9385539 - cg00455876 27653438
## cg01707559 chrY 6778695 + cg01707559 45652402
## AddressB ProbeSeqA
## <character> <character>
## cg00050873 31717405 ACAAAAAAACAACACACAACTATAATAATTTTTAAAATAAATAAACCCCA
## cg00212031 38703326 CCCAATTAACCACAAAAACTAAACAAATTATACAATCAAAAAAACATACA
## cg00213748 36767301 TTTTAACACCTAACACCATTTTAACAATAAAAATTCTACAAAAAAAAACA
## cg00214611 46723459 CTAACTTCCAAACCACACTTTATATACTAAACTACAATATAACACAAACA
## cg00455876 69732350 AACTCTAAACTACCCAACACAAACTCCAAAAACTTCTCAAAAAAAACTCA
## cg01707559 64689504 ACAAATTAAAAACACTAAAACAAACACAACAACTACAACAACAAAAAACA
## ProbeSeqB Type
## <character> <character>
## cg00050873 ACGAAAAAACAACGCACAACTATAATAATTTTTAAAATAAATAAACCCCG I
## cg00212031 CCCAATTAACCGCAAAAACTAAACAAATTATACGATCGAAAAAACGTACG I
## cg00213748 TTTTAACGCCTAACACCGTTTTAACGATAAAAATTCTACAAAAAAAAACG I
## cg00214611 CTAACTTCCGAACCGCGCTTTATATACTAAACTACAATATAACGCGAACG I
## cg00455876 AACTCTAAACTACCCGACACAAACTCCAAAAACTTCTCGAAAAAAACTCG I
## cg01707559 GCGAATTAAAAACACTAAAACGAACGCGACGACTACAACGACAAAAAACG I
## NextBase Color Probe_rs Probe_maf CpG_rs
## <character> <character> <character> <numeric> <character>
## cg00050873 A Red NA NA NA
## cg00212031 T Red NA NA NA
## cg00213748 A Red NA NA NA
## cg00214611 A Red NA NA NA
## cg00455876 A Red NA NA NA
## cg01707559 A Red NA NA NA
## CpG_maf SBE_rs SBE_maf Islands_Name
## <numeric> <character> <numeric> <character>
## cg00050873 NA NA NA chrY:9363680-9363943
## cg00212031 NA NA NA chrY:21238448-21240005
## cg00213748 NA NA NA chrY:8147877-8148210
## cg00214611 NA NA NA chrY:15815488-15815779
## cg00455876 NA NA NA chrY:9385471-9385777
## cg01707559 NA NA NA chrY:6778574-6780028
## Relation_to_Island
## <character>
## cg00050873 N_Shore
## cg00212031 Island
## cg00213748 S_Shore
## cg00214611 Island
## cg00455876 Island
## cg01707559 Island
## Forward_Sequence
## <character>
## cg00050873 TATCTCTGTCTGGCGAGGAGGCAACGCACAACTGTGGTGGTTTTTGGAGTGGGTGGACCC[CG]GCCAAGACGGCCTGGGCTGACCAGAGACGGGAGGCAGAAAAAGTGGGCAGGTGGTTGCAG
## cg00212031 CCATTGGCCCGCCCCAGTTGGCCGCAGGGACTGAGCAAGTTATGCGGTCGGGAAGACGTG[CG]TTAAAGGGCTGAAGGGGAGGGACGGAACTGACAGTCTCTGTGACAGCTCTGAGGTGGGAG
## cg00213748 TCTGTGGGACCATTTTAACGCCTGGCACCGTTTTAACGATGGAGGTTCTGCAGGAGGGGG[CG]ACCTGGGGTAGGAGGCGTGCTAGTGGTGGATGACATTGTGGCAGAGATGGAGGTGGTGGC
## cg00214611 GCGCCGGCAGGACTAGCTTCCGGGCCGCGCTTTGTGTGCTGGGCTGCAGTGTGGCGCGGG[CG]AGGAAGCTGGTAGGGCGGTTGTCGCAAGCTCCAGCTGCAGCCTCCGCCTACGTGAGAAGA
## cg00455876 CGCGTGTGCCTGGACTCTGAGCTACCCGGCACAAGCTCCAAGGGCTTCTCGGAGGAGGCT[CG]GGGACGGAAGGCGTGGGGTGAGTGGGCTGGAGATGCAGGCGCGCCCGTGGCTGTGCAGCC
## cg01707559 AGCGGCCGCTCCCAGTGGTGGTCACCGCCAGTGCCAATCCCTTGCGCCGCCGTGCAGTCC[CG]CCCTCTGTCGCTGCAGCCGCCGCGCCCGCTCCAGTGCCCCCAATTCGCGCTCGGGAGTGA
## SourceSeq Random_Loci
## <character> <character>
## cg00050873 CGGGGTCCACCCACTCCAAAAACCACCACAGTTGTGCGTTGCCTCCTCGC
## cg00212031 CGCACGTCTTCCCGACCGCATAACTTGCTCAGTCCCTGCGGCCAACTGGG
## cg00213748 CGCCCCCTCCTGCAGAACCTCCATCGTTAAAACGGTGCCAGGCGTTAAAA
## cg00214611 CGCCCGCGCCACACTGCAGCCCAGCACACAAAGCGCGGCCCGGAAGCTAG
## cg00455876 GACTCTGAGCTACCCGGCACAAGCTCCAAGGGCTTCTCGGAGGAGGCTCG
## cg01707559 CGCCCTCTGTCGCTGCAGCCGCCGCGCCCGCTCCAGTGCCCCCAATTCGC
## Methyl27_Loci UCSC_RefGene_Name UCSC_RefGene_Accession
## <character> <character> <character>
## cg00050873 TSPY4;FAM197Y2 NM_001164471;NR_001553
## cg00212031 TTTY14 NR_001543
## cg00213748
## cg00214611 TMSB4Y;TMSB4Y NM_004202;NM_004202
## cg00455876
## cg01707559 TBL1Y;TBL1Y;TBL1Y NM_134259;NM_033284;NM_134258
## UCSC_RefGene_Group Phantom DMR Enhancer
## <character> <character> <character> <character>
## cg00050873 Body;TSS1500
## cg00212031 TSS200
## cg00213748
## cg00214611 1stExon;5'UTR
## cg00455876
## cg01707559 TSS200;TSS200;TSS200
## HMM_Island Regulatory_Feature_Name
## <character> <character>
## cg00050873 Y:9973136-9976273
## cg00212031 Y:19697854-19699393
## cg00213748 Y:8207555-8208234
## cg00214611 Y:14324883-14325218 Y:15815422-15815706
## cg00455876 Y:9993394-9995882
## cg01707559 Y:6838022-6839951
## Regulatory_Feature_Group DHS
## <character> <character>
## cg00050873
## cg00212031
## cg00213748
## cg00214611 Promoter_Associated_Cell_type_specific
## cg00455876
## cg01707559The simplest way to read the raw methylation data into R is using the minfi function read.450k.sheet, along with the path to the IDAT files and a sample sheet. The sample sheet is a CSV (comma-separated) file containing one line per sample, with a number of columns describing each sample. The format expected by the read.450k.sheet function is based on the sample sheet file that usually accompanies Illumina methylation array data. It is also very similar to the targets file described by the limma package. Reading the sample sheet into R creates a data.frame with one row for each sample and several columns. The read.450k.sheet function uses the specified path and other information from the sample sheet to create a column called Basename which specifies the location of each individual IDAT file in the experiment.
# set up a path for your project projectDirectory <- "/absolute/path/to/your/project"
# set up a path to your data directory - which should be in your project directory dataDirectory <- paste(projectDirectory,"data",sep="/") # read in the sample sheet for the experiment targets <- read.450k.sheet(dataDirectory, pattern="SampleSheet.csv")
## [read.450k.sheet] Found the following CSV files:
## [1] "/group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/SampleSheet.csv"targets## Sample_Name Sample_Well Sample_Source Sample_Group Sample_Label
## 1 1 A1 M28 naive naive
## 2 2 B1 M28 rTreg rTreg
## 3 3 C1 M28 act_naive act_naive
## 4 4 D1 M29 naive naive
## 5 5 E1 M29 act_naive act_naive
## 6 6 F1 M29 act_rTreg act_rTreg
## 7 7 G1 M30 naive naive
## 8 8 H1 M30 rTreg rTreg
## 9 9 A2 M30 act_naive act_naive
## 10 10 B2 M30 act_rTreg act_rTreg
## 11 11 H06 VICS-72098-18-B birth birth
## Pool_ID Array Slide
## 1 NA R01C01 6264509100
## 2 NA R02C01 6264509100
## 3 NA R03C01 6264509100
## 4 NA R04C01 6264509100
## 5 NA R05C01 6264509100
## 6 NA R06C01 6264509100
## 7 NA R01C02 6264509100
## 8 NA R02C02 6264509100
## 9 NA R03C02 6264509100
## 10 NA R04C02 6264509100
## 11 NA R06C02 5975827018
## Basename
## 1 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R01C01
## 2 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R02C01
## 3 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R03C01
## 4 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R04C01
## 5 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R05C01
## 6 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R06C01
## 7 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R01C02
## 8 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R02C02
## 9 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R03C02
## 10 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/6264509100/6264509100_R04C02
## 11 /group/bioi1/shared/BioinfoSummer2015/450kAnalysisWorkshop/data/5975827018/5975827018_R06C02Now that we have imported the information about the samples and where the data is located, we can read the raw intensity signals into R from the IDAT files. This creates an RGChannelSet object that contains all the raw intensity data, from both the red and green colour channels, for each of the samples. At this stage, it can be useful to rename the samples with more descriptive names.
# read in the raw data from the IDAT files rgSet <- read.450k.exp(targets=targets) rgSet
## RGChannelSet (storageMode: lockedEnvironment)
## assayData: 622399 features, 11 samples
## element names: Green, Red
## An object of class 'AnnotatedDataFrame'
## sampleNames: 6264509100_R01C01 6264509100_R02C01 ...
## 5975827018_R06C02 (11 total)
## varLabels: Sample_Name Sample_Well ... filenames (10 total)
## varMetadata: labelDescription
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19# give the samples descriptive names targets$ID <- paste(targets$Sample_Group,targets$Sample_Name,sep=".") sampleNames(rgSet) <- targets$ID rgSet
## RGChannelSet (storageMode: lockedEnvironment)
## assayData: 622399 features, 11 samples
## element names: Green, Red
## An object of class 'AnnotatedDataFrame'
## sampleNames: naive.1 rTreg.2 ... birth.11 (11 total)
## varLabels: Sample_Name Sample_Well ... filenames (10 total)
## varMetadata: labelDescription
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19Once the data has been imported into R, we can evaluate its quality. Firstly, we need to calculate detection p-values. We can generate a detection p-value for every CpG in every sample, which is indicative of the quality of the signal. The method used by minfi to calculate detection p-values compares the total signal (M + U) for each probe to the background signal level, which is estimated from the negative control probes. Very small p-values are indicative of a reliable signal whilst large p-values, for example >0.01, generally indicate a poor quality signal.
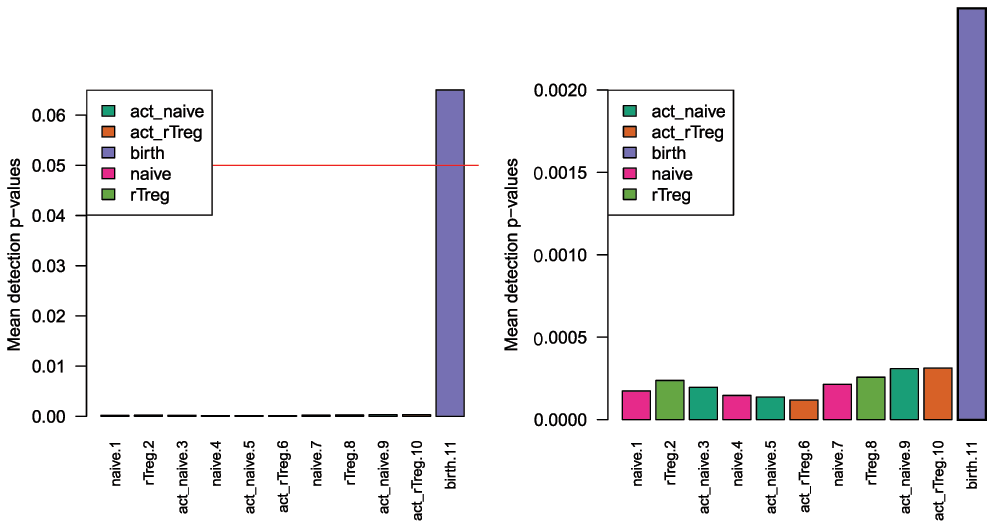
Plotting the mean detection p-value for each sample allows us to gauge the general quality of the samples in terms of the overall signal reliability (Figure 2). Samples that have many failed probes will have relatively large mean detection p-values.
# calculate the detection p-values detP <- detectionP(rgSet) head(detP)
## naive.1 rTreg.2 act_naive.3 naive.4 act_naive.5 act_rTreg.6
## cg00050873 0 0 0.000000e+00 0 0.00000e+00 0
## cg00212031 0 0 0.000000e+00 0 0.00000e+00 0
## cg00213748 0 0 1.181832e-12 0 8.21565e-15 0
## cg00214611 0 0 0.000000e+00 0 0.00000e+00 0
## cg00455876 0 0 0.000000e+00 0 0.00000e+00 0
## cg01707559 0 0 0.000000e+00 0 0.00000e+00 0
## naive.7 rTreg.8 act_naive.9 act_rTreg.10 birth.11
## cg00050873 0 0.000000e+00 0 0.000000e+00 0.0000000
## cg00212031 0 0.000000e+00 0 0.000000e+00 0.0000000
## cg00213748 0 1.469801e-05 0 1.365951e-08 0.6735224
## cg00214611 0 0.000000e+00 0 0.000000e+00 0.7344451
## cg00455876 0 0.000000e+00 0 0.000000e+00 0.0000000
## cg01707559 0 0.000000e+00 0 0.000000e+00 0.0000000# examine mean detection p-values across all samples to identify any failed samples pal <- brewer.pal(8,"Dark2") par(mfrow=c(1,2)) barplot(colMeans(detP), col=pal[factor(targets$Sample_Group)], las=2, cex.names=0.8, ylab="Mean detection p-values") abline(h=0.05,col="red") legend("topleft", legend=levels(factor(targets$Sample_Group)), fill=pal, bg="white") barplot(colMeans(detP), col=pal[factor(targets$Sample_Group)], las=2, cex.names=0.8, ylim=c(0,0.002), ylab="Mean detection p-values") abline(h=0.05,col="red") legend("topleft", legend=levels(factor(targets$Sample_Group)), fill=pal, bg="white")
The minfi qcReport function generates many other useful quality control plots. The minfi vignette describes the various plots and how they should be interpreted in detail. Generally, samples that look poor based on mean detection p-value will also look poor using other metrics and it is usually advisable to exclude them from further analysis.
qcReport(rgSet, sampNames=targets$ID, sampGroups=targets$Sample_Group, pdf="qcReport.pdf")
Poor quality samples can be easily excluded from the analysis using a detection p-value cutoff, for example >0.05. For this particular dataset, the birth sample shows a very high mean detection p-value, and hence it is excluded from subsequent analysis (Figure 2).
# remove poor quality samples keep <- colMeans(detP) < 0.05 rgSet <- rgSet[,keep] rgSet
## RGChannelSet (storageMode: lockedEnvironment)
## assayData: 622399 features, 10 samples
## element names: Green, Red
## An object of class 'AnnotatedDataFrame'
## sampleNames: naive.1 rTreg.2 ... act_rTreg.10 (10 total)
## varLabels: Sample_Name Sample_Well ... filenames (10 total)
## varMetadata: labelDescription
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19# remove poor quality samples from targets data targets <- targets[keep,] targets[,1:5]
## Sample_Name Sample_Well Sample_Source Sample_Group Sample_Label
## 1 1 A1 M28 naive naive
## 2 2 B1 M28 rTreg rTreg
## 3 3 C1 M28 act_naive act_naive
## 4 4 D1 M29 naive naive
## 5 5 E1 M29 act_naive act_naive
## 6 6 F1 M29 act_rTreg act_rTreg
## 7 7 G1 M30 naive naive
## 8 8 H1 M30 rTreg rTreg
## 9 9 A2 M30 act_naive act_naive
## 10 10 B2 M30 act_rTreg act_rTreg# remove poor quality samples from detection p-value table detP <- detP[,keep] dim(detP)
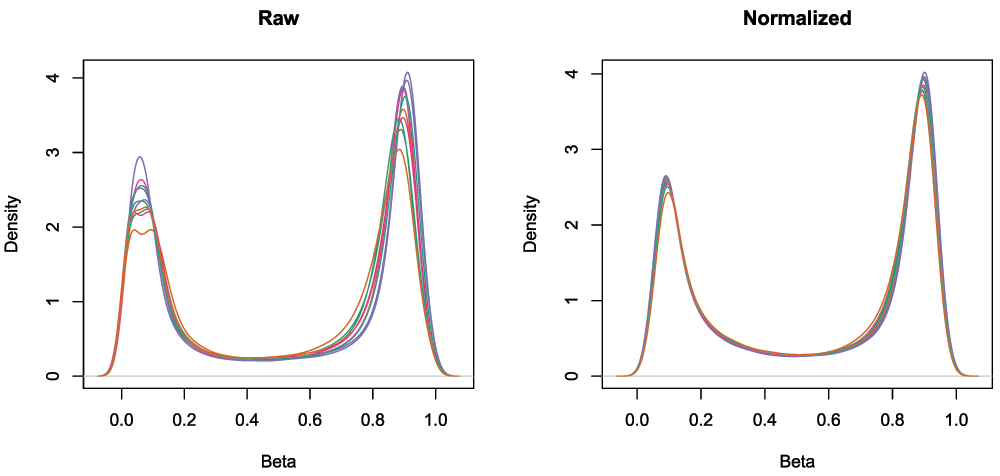
## [1] 485512 10To minimise the unwanted variation within and between samples, various data normalizations can be applied. Many different types of normalization have been developed for methylation arrays and it is beyond the scope of this workflow to compare and contrast all of them (Fortin et al., 2014; Maksimovic et al., 2012; Mancuso et al., 2011; Pidsley et al., 2013; Sun et al., 2011; Teschendorff et al., 2013; Touleimat & Tost, 2012; Triche et al., 2013; Wang et al., 2012; Wu et al., 2014). Several methods have been built into minfi and can be directly applied within its framework (Fortin et al., 2014; Maksimovic et al., 2012; Touleimat & Tost, 2012; Triche et al., 2013), whilst others are methylumi-specific or require custom data types (Mancuso et al., 2011; Pidsley et al., 2013; Sun et al., 2011; Teschendorff et al., 2013; Wang et al., 2012; Wu et al., 2014). Although there is no single normalisation method that is universally considered best, a recent study by Fortin et al. (2014) has suggested that a good rule of thumb within the minfi framework is that the preprocessFunnorm (Fortin et al., 2014) function is most appropriate for datasets with global methylation differences such as cancer/normal or vastly different tissue types, whilst the preprocessQuantile function (Touleimat & Tost, 2012) is more suited for datasets where you do not expect global differences between your samples, for example a single tissue. As we are comparing different blood cell types, which are globally relatively similar, we will apply the preprocessQuantile method to our data (Figure 3). Note that after normalization, the data is housed in a GenomicRatioSet object. This is a much more compact representation of the data as the colour channel information has been discarded and the M and U intensity information has been converted to M-values and beta values, together with associated genomic coordinates.
# normalize the data; this results in a GenomicRatioSet object mSetSq <- preprocessQuantile(rgSet)
## [preprocessQuantile] Mapping to genome.
## [preprocessQuantile] Fixing outliers.## Warning in .getSex(CN = CN, xIndex = xIndex, yIndex = yIndex, cutoff
## = cutoff): An inconsistency was encountered while determining sex. One
## possibility is that only one sex is present. We recommend further checks,
## for example with the plotSex function.## [preprocessQuantile] Quantile normalizing.# create a MethylSet object from the raw data for plotting mSetRaw <- preprocessRaw(rgSet)
# visualise what the data looks like before and after normalization par(mfrow=c(1,2)) densityPlot(rgSet, sampGroups=targets$Sample_Group,main="Raw", legend=FALSE) densityPlot(getBeta(mSetSq), sampGroups=targets$Sample_Group, main="Normalized", legend=FALSE)
Multi-dimensional scaling (MDS) plots are excellent for visualising data, and are usually some of the first plots that should be made when exploring the data. MDS plots are based on principle components analysis and are an unsupervised method for looking at the similarities and differences between the various samples. Samples that are more similar to each other should cluster together, and samples that are very different should be further apart on the plot. Dimension one (or principle component one) captures the greatest source of variation in the data, dimension two captures the second greatest source of variation in the data and so on. Colouring the data points or labels by known factors of interest can often highlight exactly what the greatest sources of variation are in the data. It is also possible to use MDS plots to decipher sample mix-ups.
# MDS plots to look at largest sources of variation par(mfrow=c(1,2)) plotMDS(getM(mSetSq), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)]) legend("top", legend=levels(factor(targets$Sample_Group)), text.col=pal, bg="white", cex=0.7) plotMDS(getM(mSetSq), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Source)]) legend("top", legend=levels(factor(targets$Sample_Source)), text.col=pal, bg="white", cex=0.7)
Examining the MDS plots for this dataset demonstrates that the largest source of variation is the difference between individuals (Figure 4). The higher dimensions reveal that the differences between cell types are largely captured by the third and fourth principal components (Figure 5). This type of information is useful in that it can inform downstream analysis by including obvious sources of unwanted variation in our statistical model to account for them, in this case individual to individual variation.
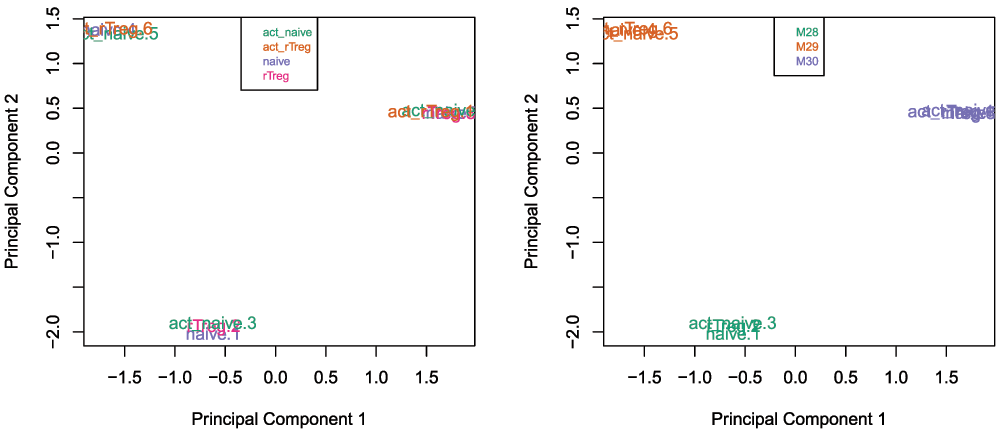
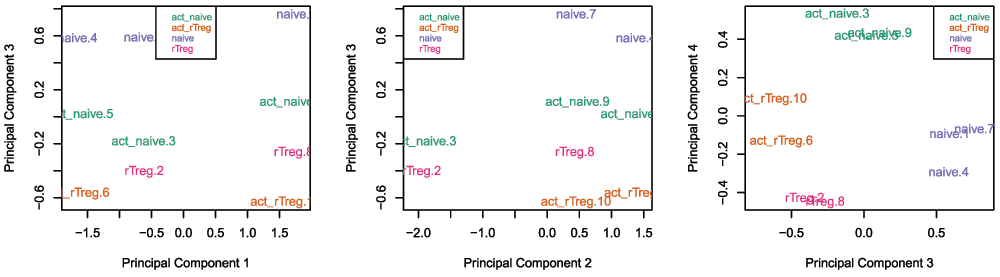
# Examine higher dimensions to look at other sources of variation par(mfrow=c(1,3)) plotMDS(getM(mSetSq), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)], dim=c(1,3)) legend("top", legend=levels(factor(targets$Sample_Group)), text.col=pal, cex=0.7, bg="white") plotMDS(getM(mSetSq), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)], dim=c(2,3)) legend("topleft", legend=levels(factor(targets$Sample_Group)), text.col=pal, cex=0.7, bg="white") plotMDS(getM(mSetSq), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)], dim=c(3,4)) legend("topright", legend=levels(factor(targets$Sample_Group)), text.col=pal, cex=0.7, bg="white")
Poor performing probes are generally filtered out prior to differential methylation analysis. As the signal from these probes is unreliable, by removing them we perform fewer statistical tests and thus incur a reduced multiple testing penalty. We filter out probes that have failed in one or more samples based on detection p-value.
# ensure probes are in the same order in the mSetSq and detP objects detP <- detP[match(featureNames(mSetSq),rownames(detP)),]
# remove any probes that have failed in one or more samples keep <- rowSums(detP < 0.01) == ncol(mSetSq) table(keep)
## keep
## FALSE TRUE
## 977 484535mSetSqFlt <- mSetSq[keep,]
mSetSqFlt## class: GenomicRatioSet
## dim: 484535 10
## metadata(0):
## assays(2): M CN
## rownames(484535): cg13869341 cg14008030 ... cg08265308 cg14273923
## rowRanges metadata column names(0):
## colnames(10): naive.1 rTreg.2 ... act_naive.9 act_rTreg.10
## colData names(11): Sample_Name Sample_Well ... filenames
## predictedSex
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19
## Preprocessing
## Method: Raw (no normalization or bg correction)
## minfi version: 1.16.1
## Manifest version: 0.4.0Depending on the nature of your samples and your biological question you may also choose to filter out the probes from the X and Y chromosomes or probes that are known to have common SNPs at the CpG site. As the samples in this dataset were all derived from male donors, we will not be removing the sex chromosome probes as part of this analysis, however example code is provided below. A different dataset, which contains both male and female samples, is used to demonstrate a Differential Variability analysis and provides an example of when sex chromosome removal is necessary (Figure 13).
# if your data includes males and females, remove probes on the sex chromosomes keep <- !(featureNames(mSetSqFlt) %in% ann450k$Name[ann450k$chr %in% c("chrX","chrY")]) table(keep) mSetSqFlt <- mSetSqFlt[keep,]
There is a function in minfi that provides a simple interface for the removal of probes where common SNPs may affect the CpG. You can either remove all probes affected by SNPs (default), or only those with minor allele frequencies greater than a specified value.
# remove probes with SNPs at CpG site mSetSqFlt <- dropLociWithSnps(mSetSqFlt) mSetSqFlt
## class: GenomicRatioSet
## dim: 467351 10
## metadata(0):
## assays(2): M CN
## rownames(467351): cg13869341 cg14008030 ... cg08265308 cg14273923
## rowRanges metadata column names(0):
## colnames(10): naive.1 rTreg.2 ... act_naive.9 act_rTreg.10
## colData names(11): Sample_Name Sample_Well ... filenames
## predictedSex
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19
## Preprocessing
## Method: Raw (no normalization or bg correction)
## minfi version: 1.16.1
## Manifest version: 0.4.0We will also filter out probes that have shown to be cross-reactive, that is, probes that have been demonstrated to map to multiple places in the genome. This list was originally published by Chen et al. (2013) and can be obtained from the authors’ website.
# exclude cross reactive probes xReactiveProbes <- read.csv(file=paste(dataDirectory, "48639-non-specific-probes-Illumina450k.csv", sep="/"), stringsAsFactors=FALSE) keep <- !(featureNames(mSetSqFlt) %in% xReactiveProbes$TargetID) table(keep)
## keep
## FALSE TRUE
## 27433 439918mSetSqFlt <- mSetSqFlt[keep,]
mSetSqFlt## class: GenomicRatioSet
## dim: 439918 10
## metadata(0):
## assays(2): M CN
## rownames(439918): cg13869341 cg24669183 ... cg08265308 cg14273923
## rowRanges metadata column names(0):
## colnames(10): naive.1 rTreg.2 ... act_naive.9 act_rTreg.10
## colData names(11): Sample_Name Sample_Well ... filenames
## predictedSex
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19
## Preprocessing
## Method: Raw (no normalization or bg correction)
## minfi version: 1.16.1
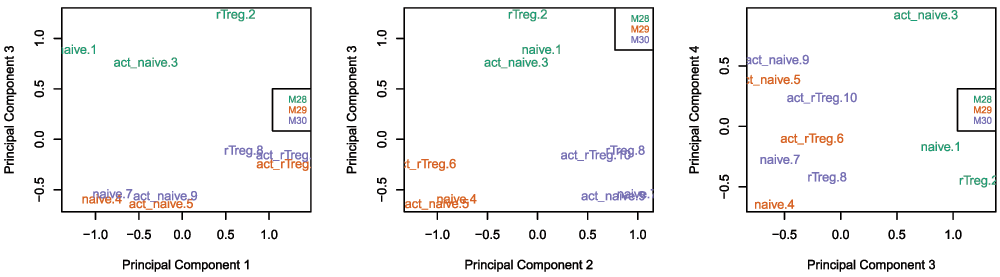
## Manifest version: 0.4.0Once the data has been filtered and normalised, it is often useful to re-examine the MDS plots to see if the relationship between the samples has changed. It is apparent from the new MDS plots that much of the inter-individual variation has been removed as this is no longer the first principal component (Figure 6), likely due to the removal of the SNP-affected CpG probes. However, the samples do still cluster by individual in the second dimension (Figure 6 and Figure 7) and thus a factor for individual should still be included in the model.
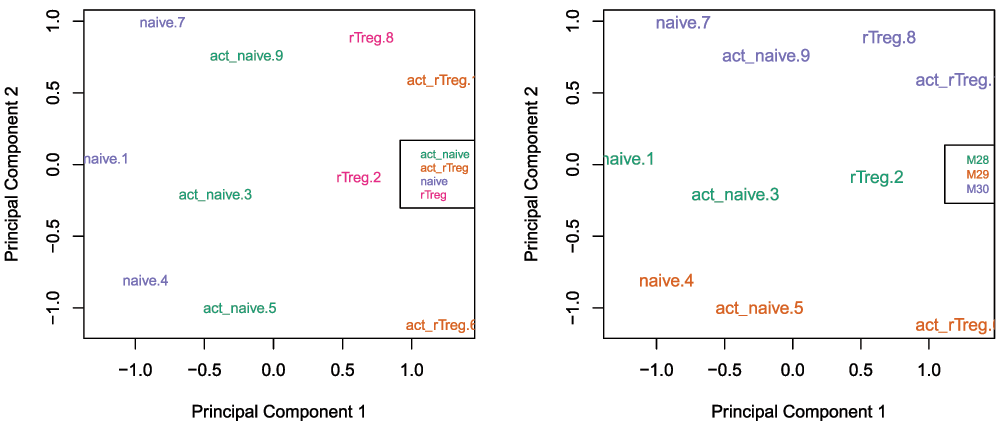
par(mfrow=c(1,2)) plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)], cex=0.8) legend("right", legend=levels(factor(targets$Sample_Group)), text.col=pal, cex=0.65, bg="white") plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Source)]) legend("right", legend=levels(factor(targets$Sample_Source)), text.col=pal, cex=0.7, bg="white")
par(mfrow=c(1,3)) # Examine higher dimensions to look at other sources of variation plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Source)], dim=c(1,3)) legend("right", legend=levels(factor(targets$Sample_Source)), text.col=pal, cex=0.7, bg="white") plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Source)], dim=c(2,3)) legend("topright", legend=levels(factor(targets$Sample_Source)), text.col=pal, cex=0.7, bg="white") plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Source)], dim=c(3,4)) legend("right", legend=levels(factor(targets$Sample_Source)), text.col=pal, cex=0.7, bg="white")
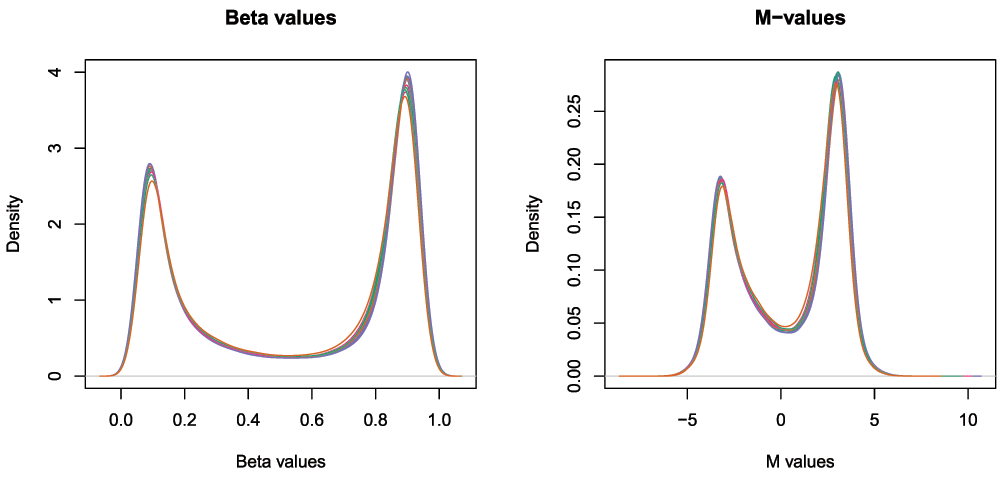
The next step is to calculate M-values and beta values (Figure 8). As previously mentioned, M-values have nicer statistical properties and are thus better for use in statistical analysis of methylation data whilst beta values are easy to interpret and are thus better for displaying data. A detailed comparison of M-values and beta values was published by Du et al. (2010).
# calculate M-values for statistical analysis mVals <- getM(mSetSqFlt) head(mVals[,1:5])
## naive.1 rTreg.2 act_naive.3 naive.4 act_naive.5
## cg13869341 2.421276 2.515948 2.165745 2.286314 2.109441
## cg24669183 2.169414 2.235964 2.280734 1.632309 2.184435
## cg15560884 1.761176 1.577578 1.597503 1.777486 1.764999
## cg01014490 -3.504268 -3.825119 -5.384735 -4.537864 -4.296526
## cg17505339 3.082191 3.924931 4.163206 3.255373 3.654134
## cg11954957 1.546401 1.912204 1.727910 2.441267 1.618331bVals <- getBeta(mSetSqFlt) head(bVals[,1:5])
## naive.1 rTreg.2 act_naive.3 naive.4 act_naive.5
## cg13869341 0.84267937 0.85118462 0.8177504 0.82987650 0.81186174
## cg24669183 0.81812908 0.82489238 0.8293297 0.75610281 0.81967323
## cg15560884 0.77219626 0.74903910 0.7516263 0.77417882 0.77266205
## cg01014490 0.08098986 0.06590459 0.0233755 0.04127262 0.04842397
## cg17505339 0.89439216 0.93822870 0.9471357 0.90520570 0.92641305
## cg11954957 0.74495496 0.79008516 0.7681146 0.84450764 0.75431167par(mfrow=c(1,2)) densityPlot(bVals, sampGroups=targets$Sample_Group, main="Beta values", legend=FALSE, xlab="Beta values") densityPlot(mVals, sampGroups=targets$Sample_Group, main="M-values", legend=FALSE, xlab="M values")
The biological question of interest for this particular dataset is to discover differentially methylated probes between the different cell types. However, as was apparent in the MDS plots, there is another factor that we need to take into account when we perform the statistical analysis. In the targets file, there is a column called Sample_Source, which refers to the individuals that the samples were collected from. In this dataset, each of the individuals contributes more than one cell type. For example, individual M28 contributes naive, rTreg and act_naive samples. Hence, when we specify our design matrix, we need to include two factors: individual and cell type. This style of analysis is called a paired analysis; differences between cell types are calculated within each individual, and then these differences are averaged across individuals to determine whether there is an overall significant difference in the mean methylation level for each CpG site. The limma User’s Guide extensively covers the different types of designs that are commonly used for microarray experiments and how to analyse them in R.
We are interested in pairwise comparisons between the four cell types, taking into account individual to individual variation. We perform this analysis on the matrix of M-values in limma, obtaining moderated t-statistics and associated p-values for each CpG site. The comparison that has the most significantly differentially methylated CpGs is naive vs rTreg (n=3021 at 5% false discovery rate (FDR)), while rTreg vs act_rTreg doesn’t show any significant differential methylation.
# this is the factor of interest cellType <- factor(targets$Sample_Group) # this is the individual effect that we need to account for individual <- factor(targets$Sample_Source) # use the above to create a design matrix design <- model.matrix(~0+cellType+individual, data=targets) colnames(design) <- c(levels(cellType),levels(individual)[-1]) # fit the linear model fit <- lmFit(mVals, design) # create a contrast matrix for specific comparisons contMatrix <- makeContrasts(naive-rTreg, naive-act_naive, rTreg-act_rTreg, act_naive-act_rTreg, levels=design) contMatrix
## Contrasts
## Levels naive - rTreg naive - act_naive rTreg - act_rTreg
## act_naive 0 -1 0
## act_rTreg 0 0 -1
## naive 1 1 0
## rTreg -1 0 1
## M29 0 0 0
## M30 0 0 0
## Contrasts
## Levels act_naive - act_rTreg
## act_naive 1
## act_rTreg -1
## naive 0
## rTreg 0
## M29 0
## M30 0# fit the contrasts fit2 <- contrasts.fit(fit, contMatrix) fit2 <- eBayes(fit2) # look at the numbers of DM CpGs at FDR < 0.05 summary(decideTests(fit2))
## naive - rTreg naive - act_naive rTreg - act_rTreg act_naive - act_rTreg
## -1 1618 400 0 559
## 0 436897 439291 439918 438440
## 1 1403 227 0 919We can extract the tables of differentially expressed CpGs for each comparison, ordered by B-statistic by default, using the topTable function in limma. The results of the analysis for the first comparison, naive vs. rTreg, can be saved as a data.frame by setting coef=1.
# get the table of results for the first contrast (naive - rTreg) ann450kSub <- ann450k[match(rownames(mVals),ann450k$Name), c(1:4,12:19,24:ncol(ann450k))] DMPs <- topTable(fit2, num=Inf, coef=1, genelist=ann450kSub) head(DMPs)
## chr pos strand Name Probe_rs Probe_maf CpG_rs
## cg07499259 chr1 12188502 + cg07499259 <NA> NA <NA>
## cg26992245 chr8 29848579 - cg26992245 <NA> NA <NA>
## cg09747445 chr15 70387268 - cg09747445 <NA> NA <NA>
## cg18808929 chr8 61825469 - cg18808929 <NA> NA <NA>
## cg25015733 chr2 99342986 - cg25015733 <NA> NA <NA>
## cg21179654 chr3 114057297 + cg21179654 <NA> NA <NA>
## CpG_maf SBE_rs SBE_maf Islands_Name
## cg07499259 NA <NA> NA
## cg26992245 NA <NA> NA
## cg09747445 NA <NA> NA chr15:70387929-70393206
## cg18808929 NA <NA> NA chr8:61822358-61823028
## cg25015733 NA <NA> NA chr2:99346882-99348177
## cg21179654 NA <NA> NA
## Relation_to_Island
## cg07499259 OpenSea
## cg26992245 OpenSea
## cg09747445 N_Shore
## cg18808929 S_Shelf
## cg25015733 N_Shelf
## cg21179654 OpenSea
## UCSC_RefGene_Name
## cg07499259 TNFRSF8;TNFRSF8
## cg26992245
## cg09747445 TLE3;TLE3;TLE3
## cg18808929
## cg25015733 MGAT4A
## cg21179654 ZBTB20;ZBTB20;ZBTB20;ZBTB20;ZBTB20;ZBTB20;ZBTB20
## UCSC_RefGene_Accession
## cg07499259 NM_152942;NM_001243
## cg26992245
## cg09747445 NM_001105192;NM_020908;NM_005078
## cg18808929
## cg25015733 NM_012214
## cg21179654 NM_001164343;NM_001164346;NM_001164345;NM_001164342;NM_001164344;NM_001164347;NM_015642
## UCSC_RefGene_Group Phantom DMR Enhancer
## cg07499259 5'UTR;Body
## cg26992245 TRUE
## cg09747445 Body;Body;Body
## cg18808929 TRUE
## cg25015733 5'UTR
## cg21179654 3'UTR;3'UTR;3'UTR;3'UTR;3'UTR;3'UTR;3'UTR
## HMM_Island Regulatory_Feature_Name
## cg07499259 1:12111023-12111225
## cg26992245
## cg09747445
## cg18808929
## cg25015733
## cg21179654 3:114057192-114057775
## Regulatory_Feature_Group DHS logFC AveExpr
## cg07499259 3.654104 2.46652171
## cg26992245 4.450696 -0.09180715
## cg09747445 -3.337299 -0.25201484
## cg18808929 -2.990263 0.77522878
## cg25015733 -3.054336 0.83280190
## cg21179654 Unclassified_Cell_type_specific 2.859016 1.32460816
## t P.Value adj.P.Val B
## cg07499259 18.73131 7.267204e-08 0.005067836 7.453206
## cg26992245 18.32674 8.615461e-08 0.005067836 7.359096
## cg09747445 -18.24438 8.923101e-08 0.005067836 7.339443
## cg18808929 -17.90181 1.034217e-07 0.005067836 7.255825
## cg25015733 -17.32615 1.333546e-07 0.005067836 7.108231
## cg21179654 17.27804 1.362674e-07 0.005067836 7.095476The resulting data.frame can easily be written to a CSV file, which can be opened in Excel.
write.table(DMPs, file="DMPs.csv", sep=",", row.names=FALSE)
It is always useful to plot sample-wise methylation levels for the top differentially methylated CpG sites to quickly ensure the results make sense (Figure 9). If the plots do not look as expected, it is usually an indication of an error in the code, or in setting up the design matrix. It is easier to interpret methylation levels on the beta value scale, so although the analysis is performed on the M-value scale, we visualise data on the beta value scale. The plotCpg function in minfi is a convenient way to plot the sample-wise beta values stratified by the grouping variable.
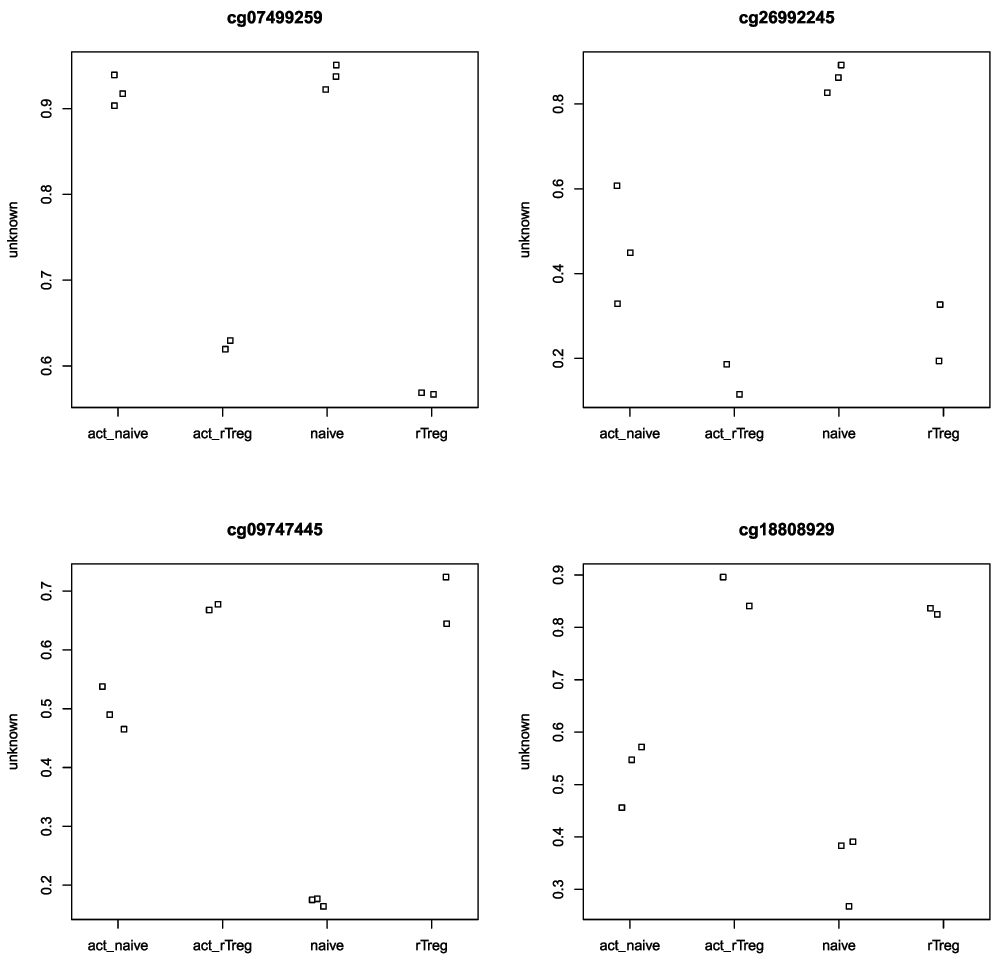
# plot the top 4 most significantly differentially methylated CpGs par(mfrow=c(2,2)) sapply(rownames(DMPs)[1:4], function(cpg){ plotCpg(bVals, cpg=cpg, pheno=targets$Sample_Group) })
## $cg07499259
## NULL
##
## $cg26992245
## NULL
##
## $cg09747445
## NULL
##
## $cg18808929
## NULLAlthough performing a probe-wise analysis is useful and informative, sometimes we are interested in knowing whether several proximal CpGs are concordantly differentially methylated, that is, we want to identify differentially methylated regions. There are several Bioconductor packages that have functions for identifying differentially methylated regions from 450k data. Some of the most popular are the dmrFind function in the charm package, which has been somewhat superseded for 450k arrays by the bumphunter function in minfi (Aryee et al., 2014; Jaffe et al., 2012), and, the recently published dmrcate in the DMRcate package (Peters et al., 2015). They are each based on different statistical methods. In our experience, the bumphunter and dmrFind functions can be somewhat slow to run unless you have the computer infrastructure to parallelise them, as they use permutations to assign significance. In this workflow, we will perform an analysis using the dmrcate. As it is based on limma, we can directly use the design and contMatrix we previously defined.
Firstly, our matrix of M-values is annotated with the relevant information about the probes such as their genomic position, gene annotation, etc. By default, this is done using the ilmn12.hg19 annotation, but this can be substituted for any argument compatible with the interface provided by the minfi package. The limma pipeline is then used for differential methylation analysis to calculate moderated t-statistics.
myAnnotation <- cpg.annotate(mVals, datatype = "array", analysis.type="differential", design=design, contrasts = TRUE, cont.matrix = contMatrix, coef="naive - rTreg")
## Your contrast returned 3021 individually significant probes. We recommend the default setting of pcutoff in dmrcate().str(myAnnotation)
## List of 6
## $ ID : Factor w/ 439918 levels "cg00000029","cg00000108",..: 232388 391918 260351 19418 289954 202723 379224
## $ stat : num [1:439918] 0.0489 -2.0773 0.7711 -0.0304 -0.764 ...
## $ CHR : Factor w/ 24 levels "chr1","chr10",..: 1 1 1 1 1 1 1 1 1 1 ...
## $ pos : int [1:439918] 15865 534242 710097 714177 720865 758829 763119 779995 805102 805338 ...
## $ betafc: num [1:439918] 0.00039 -0.04534 0.01594 0.00251 -0.00869 ...
## $ indfdr: num [1:439918] 0.994 0.565 0.872 0.997 0.873 ...
## - attr(*, "row.names")= int [1:439918] 425663 55771 233635 431055 235233 185639 266099 7424 229446 345572 ...
## - attr(*, "class")= chr "annot"Once we have the relevant statistics for the individual CpGs, we can then use the dmrcate function to combine them to identify differentially methylated regions. The main output table DMRs$results contains all of the regions found, along with their genomic annotations and p-values.
DMRs <- dmrcate(myAnnotation, lambda=1000, C=2)
## Fitting chr1...
## Fitting chr10...
## Fitting chr11...
## Fitting chr12...
## Fitting chr13...
## Fitting chr14...
## Fitting chr15...
## Fitting chr16...
## Fitting chr17...
## Fitting chr18...
## Fitting chr19...
## Fitting chr2...
## Fitting chr20...
## Fitting chr21...
## Fitting chr22...
## Fitting chr3...
## Fitting chr4...
## Fitting chr5...
## Fitting chr6...
## Fitting chr7...
## Fitting chr8...
## Fitting chr9...
## Fitting chrX...
## Fitting chrY...
## Demarcating regions...
## Done!head(DMRs$results)
## coord no.cpgs minfdr Stouffer
## 457 chr17:57915665-57918682 12 4.957890e-91 6.639928e-10
## 733 chr3:114012316-114012912 5 1.622885e-180 1.515378e-07
## 469 chr17:74639731-74640078 6 9.516873e-90 1.527961e-07
## 1069 chrX:49121205-49122718 6 6.753751e-84 2.936984e-07
## 492 chr18:21452730-21453131 7 5.702319e-115 7.674943e-07
## 186 chr10:135202522-135203200 6 1.465070e-65 7.918224e-07
## maxbetafc meanbetafc
## 457 0.3982862 0.3131611
## 733 0.5434277 0.4251622
## 469 -0.2528645 -0.1951904
## 1069 0.4529088 0.3006242
## 492 -0.3867474 -0.2546089
## 186 0.2803157 0.2293419As for the probe-wise analysis, it is advisable to visualise the results to ensure that they make sense. The regions can easily be viewed using the DMR.plot function provided in the DMRcate package (Figure 10).
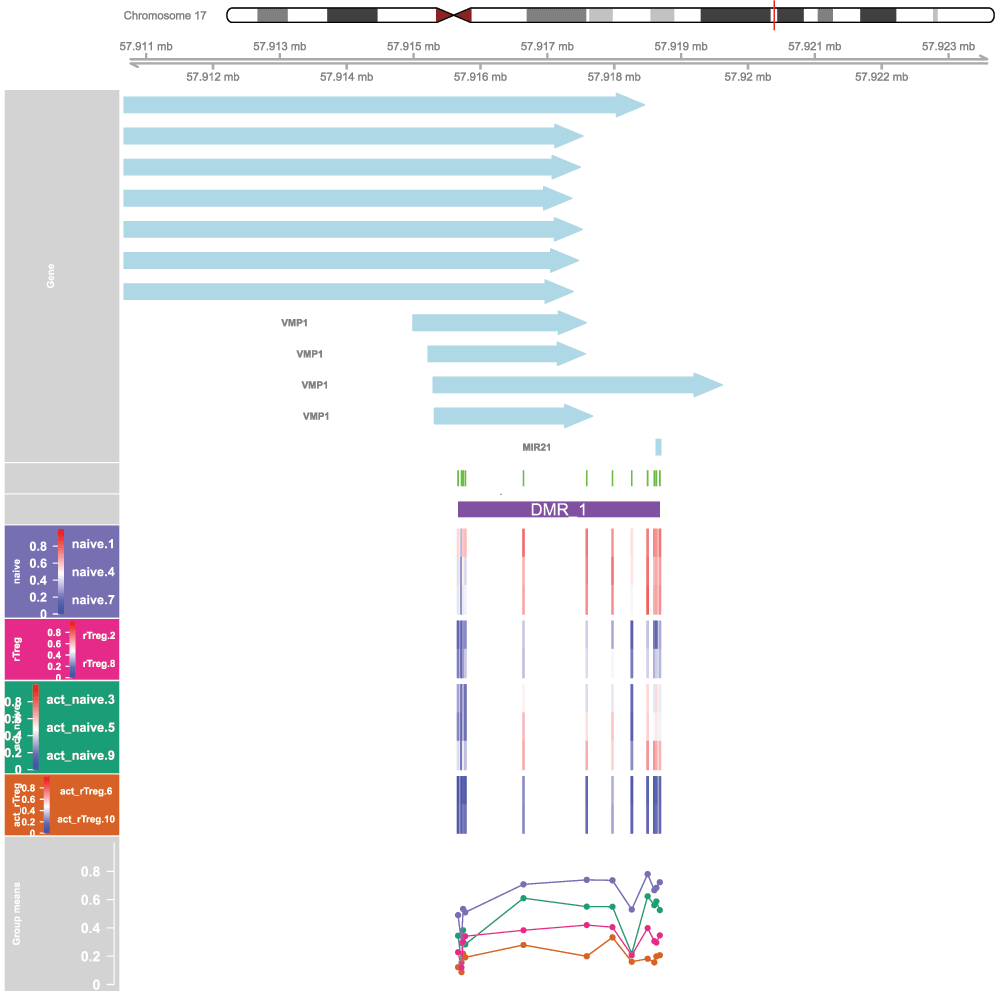
# convert the regions to annotated genomic ranges data(dmrcatedata) results.ranges <- extractRanges(DMRs, genome = "hg19") # set up the grouping variables and colours groups <- pal[1:length(unique(targets$Sample_Group))] names(groups) <- levels(factor(targets$Sample_Group)) cols <- groups[as.character(factor(targets$Sample_Group))] samps <- 1:nrow(targets)
# draw the plot par(mfrow=c(1,1)) DMR.plot(ranges=results.ranges, dmr=1, CpGs=bVals, phen.col=cols, pch=16, toscale=TRUE, plotmedians=TRUE, genome="hg19", samps=samps)
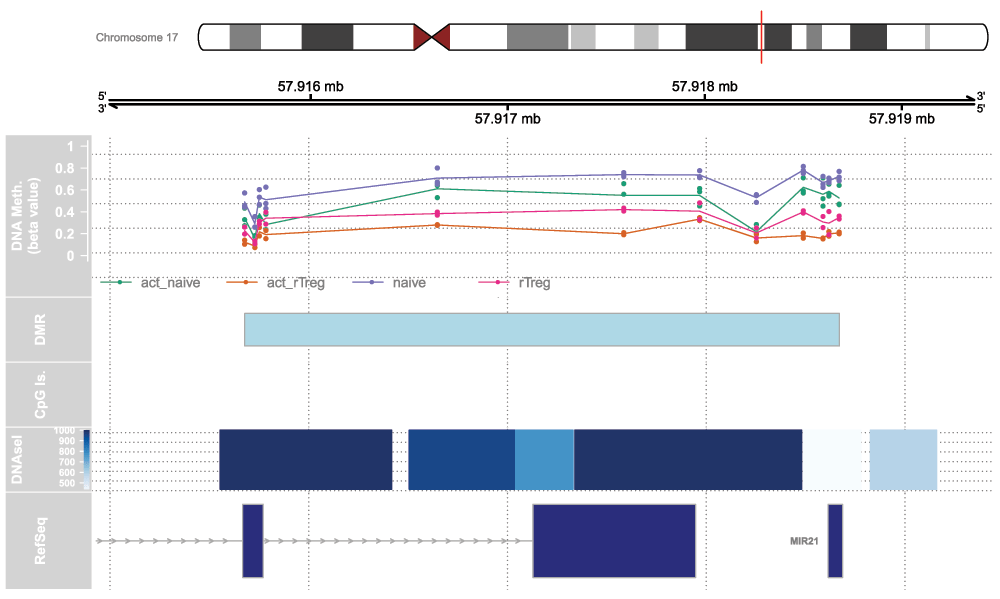
The Gviz package offers powerful functionality for plotting methylation data in its genomic context. The package vignette is very extensive and covers the various types of plots that can be produced using the Gviz framework. We will re-plot the top differentially methylated region from the DMRcate regional analysis to demonstrate the type of visualisations that can be created (Figure 11).
We will first set up the genomic region we would like to plot by extracting the genomic coordinates of the top differentially methylated region.
# indicate which genome is being used gen <- "hg19" # extract chromosome number and location from DMR results coords <- strsplit2(DMRs$results$coord[1],":") chrom <- coords[1] start <- as.numeric(strsplit2(coords[2],"-")[1]) end <- as.numeric(strsplit2(coords[2],"-")[2]) # add 25% extra space to plot minbase <- start - (0.25*(end-start)) maxbase <- end + (0.25*(end-start))
Next, we will add some genomic annotations of interest such as the locations of CpG islands and DNAseI hypersensitive sites; this can be any feature or genomic annotation of interest that you have data available for. The CpG islands data was generated using the method published by Wu et al. (2010); the DNAseI hypersensitive site data was obtained from the UCSC Genome Browser.
# CpG islands islandHMM = read.csv(paste(dataDirectory, "model-based-cpg-islands-hg19.txt", sep="/"), sep="\t", stringsAsFactors=FALSE, header=TRUE) head(islandHMM)
## chr start end length CpGcount GCcontent pctGC obsExp
## 1 chr10 93098 93818 721 32 403 0.559 0.572
## 2 chr10 94002 94165 164 12 97 0.591 0.841
## 3 chr10 94527 95302 776 65 538 0.693 0.702
## 4 chr10 119652 120193 542 53 369 0.681 0.866
## 5 chr10 122133 122621 489 51 339 0.693 0.880
## 6 chr10 180265 180720 456 32 256 0.561 0.893islandData <- GRanges(seqnames=Rle(islandHMM$chr), ranges=IRanges(start=islandHMM$start, end=islandHMM$end), strand=Rle(strand(rep("*",nrow(islandHMM))))) islandData <- islandData[seqnames(islandData) == chrom & (start(islandData) >= minbase & end(islandData) <= maxbase)] islandData
## GRanges object with 0 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## -------
## seqinfo: 81 sequences from an unspecified genome; no seqlengths# DNAseI hypersensitive sites dnase <- read.csv(paste(dataDirectory,"wgEncodeRegDnaseClusteredV3.bed", sep="/"), sep="\t",stringsAsFactors=FALSE,header=FALSE) head(dnase)
## V1 V2 V3 V4 V5 V6
## 1 chr1 10100 10330 38 261 38
## 2 chr1 10345 10590 4 310 4
## 3 chr1 16100 16315 5 158 5
## 4 chr1 65905 66055 1 157 1
## 5 chr1 91405 91615 4 278 4
## 6 chr1 115600 115790 3 545 3
## V7
## 1 3,12,13,15,21,22,32,37,36,38,39,40,50,56,57,58,59,60,53,54,62,70,76,85,93,95,103,111,117,120,1,31,77,79,73,75,87,116,
## 2 10,85,95,31,
## 3 19,13,15,46,111,
## 4 19,
## 5 9,26,72,12,
## 6 15,103,86,
## V8
## 1 50,247,129,38,52,89,138,61,54,65,35,108,198,34,68,31,48,26,59,42,109,34,105,253,56,204,99,261,101,97,19,59,88,53,72,49,46,140,
## 2 37,142,124,310,
## 3 143,158,102,33,80,
## 4 157,
## 5 172,278,223,62,
## 6 324,57,545,dnaseData <- GRanges(seqnames=dnase[,1], ranges=IRanges(start=dnase[,2], end=dnase[,3]), strand=Rle(rep("*",nrow(dnase))), data=dnase[,5]) dnaseData <- dnaseData[seqnames(dnaseData) == chrom & (start(dnaseData) >= minbase & end(dnaseData) <= maxbase)] dnaseData
## GRanges object with 6 ranges and 1 metadata column:
## seqnames ranges strand | data
## <Rle> <IRanges> <Rle> | <integer>
## [1] chr17 [57915540, 57916410] * | 1000
## [2] chr17 [57916500, 57917035] * | 954
## [3] chr17 [57917040, 57917330] * | 785
## [4] chr17 [57917340, 57918490] * | 1000
## [5] chr17 [57918500, 57918790] * | 440
## [6] chr17 [57918840, 57919175] * | 612
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengthsNow, set up the ideogram, genome and RefSeq tracks that will provide context for our methylation data.
iTrack <- IdeogramTrack(genome = gen, chromosome = chrom, name="") gTrack <- GenomeAxisTrack(col="black", cex=1, name="", fontcolor="black") rTrack <- UcscTrack(genome=gen, chromosome=chrom, track="refGene", from=minbase, to=maxbase, trackType="GeneRegionTrack", rstarts="exonStarts", rends="exonEnds", gene="name", symbol="name2", transcript="name", strand="strand", fill="darkblue",stacking="squish", name="RefSeq", showId=TRUE, geneSymbol=TRUE)
Ensure that the methylation data is ordered by chromosome and base position.
ann450kOrd <- ann450kSub[order(ann450kSub$chr,ann450kSub$pos),] head(ann450kOrd)
## DataFrame with 6 rows and 22 columns
## chr pos strand Name Probe_rs
## <character> <integer> <character> <character> <character>
## cg13869341 chr1 15865 + cg13869341 NA
## cg24669183 chr1 534242 - cg24669183 rs6680725
## cg15560884 chr1 710097 + cg15560884 NA
## cg01014490 chr1 714177 - cg01014490 NA
## cg17505339 chr1 720865 - cg17505339 NA
## cg11954957 chr1 758829 + cg11954957 rs115498424
## Probe_maf CpG_rs CpG_maf SBE_rs SBE_maf
## <numeric> <character> <numeric> <character> <numeric>
## cg13869341 NA NA NA NA NA
## cg24669183 0.108100 NA NA NA NA
## cg15560884 NA NA NA NA NA
## cg01014490 NA NA NA NA NA
## cg17505339 NA NA NA NA NA
## cg11954957 0.029514 NA NA NA NA
## Islands_Name Relation_to_Island UCSC_RefGene_Name
## <character> <character> <character>
## cg13869341 OpenSea WASH5P
## cg24669183 chr1:533219-534114 S_Shore
## cg15560884 chr1:713984-714547 N_Shelf
## cg01014490 chr1:713984-714547 Island
## cg17505339 OpenSea
## cg11954957 chr1:762416-763445 N_Shelf
## UCSC_RefGene_Accession UCSC_RefGene_Group Phantom
## <character> <character> <character>
## cg13869341 NR_024540 Body
## cg24669183
## cg15560884
## cg01014490
## cg17505339
## cg11954957
## DMR Enhancer HMM_Island Regulatory_Feature_Name
## <character> <character> <character> <character>
## cg13869341
## cg24669183 1:523025-524193
## cg15560884
## cg01014490 1:703784-704410 1:713802-715219
## cg17505339
## cg11954957
## Regulatory_Feature_Group DHS
## <character> <character>
## cg13869341
## cg24669183
## cg15560884
## cg01014490 Promoter_Associated
## cg17505339
## cg11954957bValsOrd <- bVals[match(ann450kOrd$Name,rownames(bVals)),] head(bValsOrd)
## naive.1 rTreg.2 act_naive.3 naive.4 act_naive.5
## cg13869341 0.84267937 0.85118462 0.8177504 0.82987650 0.81186174
## cg24669183 0.81812908 0.82489238 0.8293297 0.75610281 0.81967323
## cg15560884 0.77219626 0.74903910 0.7516263 0.77417882 0.77266205
## cg01014490 0.08098986 0.06590459 0.0233755 0.04127262 0.04842397
## cg17505339 0.89439216 0.93822870 0.9471357 0.90520570 0.92641305
## cg11954957 0.74495496 0.79008516 0.7681146 0.84450764 0.75431167
## act_rTreg.6 naive.7 rTreg.8 act_naive.9 act_rTreg.10
## cg13869341 0.8090798 0.8891851 0.88537940 0.90916748 0.88334231
## cg24669183 0.8187838 0.7903763 0.85304116 0.80930568 0.80979554
## cg15560884 0.7721528 0.7658623 0.75909061 0.78099397 0.78569274
## cg01014490 0.0644404 0.0245281 0.02832358 0.07740468 0.04640659
## cg17505339 0.9286016 0.8889361 0.87205348 0.90099782 0.93508348
## cg11954957 0.8116911 0.7832207 0.84929777 0.84719430 0.83350220Create the data tracks using the appropriate track type for each data type.
# create genomic ranges object from methylation data cpgData <- GRanges(seqnames=Rle(ann450kOrd$chr), ranges=IRanges(start=ann450kOrd$pos, end=ann450kOrd$pos), strand=Rle(rep("*",nrow(ann450kOrd))), betas=bValsOrd) # extract data on CpGs in DMR cpgData <- subsetByOverlaps(cpgData, results.ranges[1]) # methylation data track methTrack <- DataTrack(range=cpgData, groups=targets$Sample_Group,genome = gen, chromosome=chrom, ylim=c(-0.05,1.05), col=pal, type=c("a","p"), name="DNA Meth.\n(beta value)", background.panel="white", legend=TRUE, cex.title=0.8, cex.axis=0.8, cex.legend=0.8) # CpG island track islandTrack <- AnnotationTrack(range=islandData, genome=gen, name="CpG Is.", chromosome=chrom) # DNaseI hypersensitive site data track dnaseTrack <- DataTrack(range=dnaseData, genome=gen, name="DNAseI", type="gradient", chromosome=chrom) # DMR position data track dmrTrack <- AnnotationTrack(start=start, end=end, genome=gen, name="DMR", chromosome=chrom)
Set up the track list and indicate the relative sizes of the different tracks. Finally, draw the plot using the plotTracks function (Figure 11).
tracks <- list(iTrack, gTrack, methTrack, dmrTrack, islandTrack, dnaseTrack, rTrack) sizes <- c(2,2,5,2,2,2,3) # set up the relative sizes of the tracks plotTracks(tracks, from=minbase, to=maxbase, showTitle=TRUE, add53=TRUE, add35=TRUE, grid=TRUE, lty.grid=3, sizes=sizes, length(tracks))
Once you have performed a differential methylation analysis, there may be a very long list of significant CpG sites to interpret. One question a researcher may have is, “which gene pathways are over-represented for differentially methylated CpGs?” In some cases it is relatively straightforward to link the top differentially methylated CpGs to genes that make biological sense in terms of the cell types or samples being studied, but there may be many thousands of CpGs significantly differentially methylated. In order to gain an understanding of the biological processes that the differentially methylated CpGs may be involved in, we can perform gene ontology or KEGG pathway analysis using the gometh function in the missMethyl package (Phipson et al., 2016).
Let us consider the first comparison, naive vs rTreg, with the results of the analysis in the DMPs table. The gometh function takes as input a character vector of the names (e.g. cg20832020) of the significant CpG sites, and optionally, a character vector of all CpGs tested. This is recommended particularly if extensive filtering of the CpGs has been performed prior to analysis. For gene ontology testing (default), the user can specify collection="GO" for KEGG testing collection="KEGG". In the DMPs table, the Name column corresponds to the CpG name. We will select all CpG sites that have adjusted p-value of less than 0.05.
# Get the significant CpG sites at less than 5% FDR sigCpGs <- DMPs$Name[DMPs$adj.P.Val<0.05] # First 10 significant CpGs sigCpGs[1:10]
## [1] "cg07499259" "cg26992245" "cg09747445" "cg18808929" "cg25015733"
## [6] "cg21179654" "cg26280976" "cg16943019" "cg10898310" "cg25130381"# Total number of significant CpGs at 5% FDR length(sigCpGs)
## [1] 3021# Get all the CpG sites used in the analysis to form the background all <- DMPs$Name # Total number of CpG sites tested length(all)
## [1] 439918The gometh function takes into account the varying numbers of CpGs associated with each gene on the Illumina methylation arrays. For the 450k array, the numbers of CpGs mapping to genes can vary from as few as 1 to as many as 1200. The genes that have more CpGs associated with them will have a higher probability of being identified as differentially methylated compared to genes with fewer CpGs. We can look at this bias in the data by specifying plot=TRUE in the call to gometh (Figure 12).
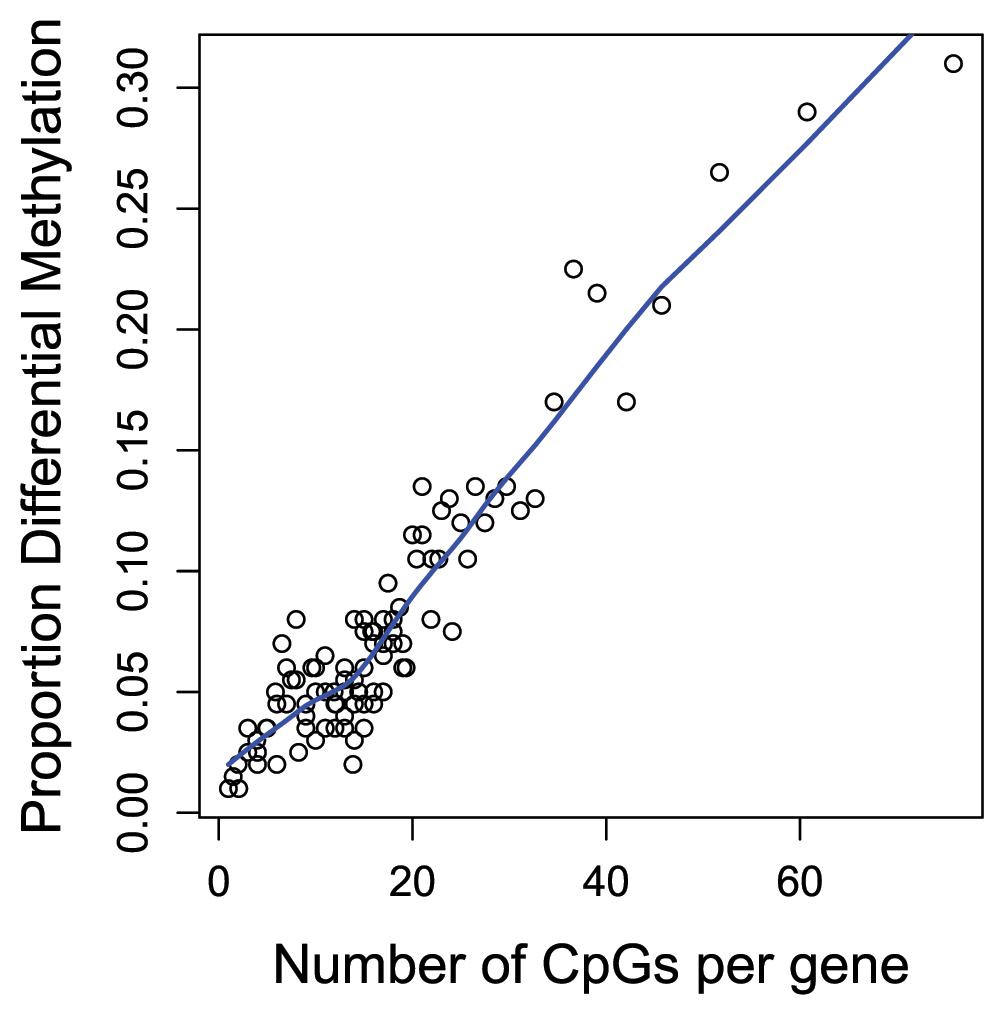
par(mfrow=c(1,1)) gst <- gometh(sig.cpg=sigCpGs, all.cpg=all, plot.bias=TRUE)
## Warning in alias2SymbolTable(flat$symbol): Multiple symbols ignored for one
## or more aliasesThe gst object is a data.frame with each row corresponding to the GO category being tested. The top 20 gene ontology categories can be displayed using the topGO function. For KEGG pathway analysis, the topKEGG function can be called to display the top 20 enriched pathways.
# Top 10 GO categories topGO(gst, number=10)
## Term Ont N DE
## GO:0002376 immune system process BP 2477 366
## GO:0007166 cell surface receptor signaling pathway BP 2613 383
## GO:0002682 regulation of immune system process BP 1435 228
## GO:0001775 cell activation BP 902 165
## GO:0007159 leukocyte cell-cell adhesion BP 451 103
## GO:0046649 lymphocyte activation BP 567 119
## GO:0045321 leukocyte activation BP 669 132
## GO:0002684 positive regulation of immune system process BP 866 154
## GO:0070486 leukocyte aggregation BP 421 97
## GO:0042110 T cell activation BP 413 95
## P.DE
## GO:0002376 0.0000000000000000000000000001390687
## GO:0007166 0.0000000000000000000072694872057477
## GO:0002682 0.0000000000000000000276016111980182
## GO:0001775 0.0000000000000000000461176043620171
## GO:0007159 0.0000000000000000000580379762162518
## GO:0046649 0.0000000000000000001374808491286637
## GO:0045321 0.0000000000000000002199145024394454
## GO:0002684 0.0000000000000000002433362861762768
## GO:0070486 0.0000000000000000008469888595049767
## GO:0042110 0.0000000000000000014907407520342597
## FDR
## GO:0002376 0.000000000000000000000002811273
## GO:0007166 0.000000000000000073476341932095
## GO:0002682 0.000000000000000185988856789313
## GO:0001775 0.000000000000000233066843044544
## GO:0007159 0.000000000000000234647537842306
## GO:0046649 0.000000000000000463195894189323
## GO:0045321 0.000000000000000614880378131680
## GO:0002684 0.000000000000000614880378131680
## GO:0070486 0.000000000000001902431088321456
## GO:0042110 0.000000000000002739574936579324From the output we can see many of the top GO categories correspond to immune system and T cell processes, which is unsurprising as the cell types being studied form part of the immune system.
For a more generalised version of gene set testing for methylation data where the user can specify the gene set to be tested, the gsameth function can be used. To display the top 20 pathways, topGSA can be called. gsameth accepts a single gene set, or a list of gene sets. The gene identifiers in the gene set must be Entrez Gene IDs. To demonstrate gsameth, we are using the curated genesets (C2) from the Broad Institute Molecular signatures database. These can be downloaded as an RData object from the WEHI Bioinformatics website.
# load Broad human curated (C2) gene sets load(paste(dataDirectory,"human_c2_v5.rdata",sep="/")) # perform the gene set test(s) gsa <- gsameth(sig.cpg=sigCpGs, all.cpg=all, collection=Hs.c2)
## Warning in alias2SymbolTable(flat$symbol): Multiple symbols ignored for one
## or more aliases# top 10 gene sets topGSA(gsa, number=10)
## N DE P.DE FDR
## REACTOME_HEMOSTASIS 466 74 0 0
## REACTOME_IMMUNE_SYSTEM 933 127 0 0
## FULCHER_INFLAMMATORY_RESPONSE_LECTIN_VS_LPS_UP 579 85 0 0
## DEURIG_T_CELL_PROLYMPHOCYTIC_LEUKEMIA_DN 320 63 0 0
## OSMAN_BLADDER_CANCER_DN 406 73 0 0
## SENESE_HDAC1_TARGETS_UP 457 71 0 0
## JAATINEN_HEMATOPOIETIC_STEM_CELL_DN 226 59 0 0
## DACOSTA_UV_RESPONSE_VIA_ERCC3_DN 855 147 0 0
## ZHANG_RESPONSE_TO_IKK_INHIBITOR_AND_TNF_UP 223 49 0 0
## HADDAD_B_LYMPHOCYTE_PROGENITOR 293 59 0 0Rather than testing for differences in mean methylation, we may be interested in testing for differences between group variances. For example, it has been hypothesised that highly variable CpGs in cancer are important for tumour progression. Hence we may be interested in CpG sites that are consistently methylated in one group, but variably methylated in another group.
Sample size is an important consideration when testing for differentially variable CpG sites. In order to get an accurate estimate of the group variances, larger sample sizes are required than for estimating group means. A good rule of thumb is to have at least ten samples in each group (Phipson & Oshlack, 2014). To demonstrate testing for differentially variable CpG sites, we will use a publicly available dataset on ageing, where whole blood samples were collected from 18 centenarians and 18 newborns and profiled for methylation on the 450k array (Heyn et al., 2012). We will first need to load, normalise and filter the data as previously described.
# set up a path to the ageing data directory age.dataDirectory <- "/absolute/path/to/your/ageing/data/directory"
age.targets <- read.450k.sheet(base=age.dataDirectory)
## [read.450k.sheet] Found the following CSV files:
## [1] "/group/bioi1/shared/public_data/ageing450k/Heyn/SampleSheet.csv"age.targets <- age.targets[age.targets$Sample_Group != "WGBS",]
# load the raw 450k from the IDAT files age.rgSet <- read.450k.exp(targets=age.targets) age.detP <- detectionP(age.rgSet) # calculate detection p-values # pre-process the data after excluding poor quality samples age.mSetSq <- preprocessQuantile(age.rgSet)
## [preprocessQuantile] Mapping to genome.
## [preprocessQuantile] Fixing outliers.
## [preprocessQuantile] Quantile normalizing.# add sex information to targets information age.targets$Sex <- getSex(age.mSetSq)$predictedSex # ensure probes are in the same order in the mSetSq and detP objects age.detP <- age.detP[match(featureNames(age.mSetSq),rownames(age.detP)),] # remove poor quality probes keep <- rowSums(age.detP < 0.01) == ncol(age.detP) age.mSetSqFlt <- age.mSetSq[keep,] # remove probes with SNPs at CpG or single base extension (SBE) site age.mSetSqFlt <- dropLociWithSnps(age.mSetSqFlt, snps = c("CpG", "SBE")) # remove cross-reactive probes keep <- !(featureNames(age.mSetSqFlt) %in% xReactiveProbes$TargetID) age.mSetSqFlt <- age.mSetSqFlt[keep,]
As this dataset contains samples from both males and females, we can use it to demonstrate the effect of removing sex chromosome probes on the data. The MDS plots below show the relationship between the samples in the ageing dataset before and after sex chromosome probe removal (Figure 13). It is apparent that before the removal of sex chromosome probes, the sample cluster based on sex in the second principal component. When the sex chromosome probes are removed, age is the largest source of variation present and the male and female samples no longer form separate clusters.
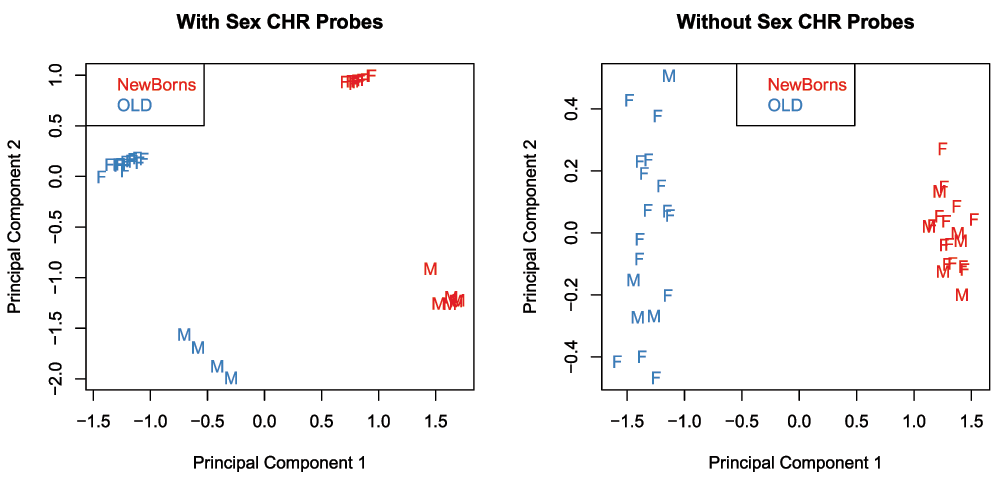
# tag sex chromosome probes for removal keep <- !(featureNames(age.mSetSqFlt) %in% ann450k$Name[ann450k$chr %in% c("chrX","chrY")]) age.pal <- brewer.pal(8,"Set1") par(mfrow=c(1,2)) plotMDS(getM(age.mSetSqFlt), top=1000, gene.selection="common", col=age.pal[factor(age.targets$Sample_Group)], labels=age.targets$Sex, main="With Sex CHR Probes") legend("topleft", legend=levels(factor(age.targets$Sample_Group)), text.col=age.pal) plotMDS(getM(age.mSetSqFlt[keep,]), top=1000, gene.selection="common", col=age.pal[factor(age.targets$Sample_Group)], labels=age.targets$Sex, main="Without Sex CHR Probes") legend("top", legend=levels(factor(age.targets$Sample_Group)), text.col=age.pal)
# remove sex chromosome probes from data age.mSetSqFlt <- age.mSetSqFlt[keep,]
We can test for differentially variable CpGs using the varFit function in the missMethyl package. The syntax for specifying which groups we are interested in testing is slightly different to the standard way a model is specified in limma, particularly for designs where an intercept is fitted (see missMethyl vignette for further details). For the ageing data, the design matrix includes an intercept term, and a term for age. The coef argument in the varFit function indicates which columns of the design matrix correspond to the intercept and grouping factor. Thus, for the ageing dataset we set coef=c(1,2). Note that design matrices without intercept terms are permitted, with specific contrasts tested using the contrasts.varFit function.
# get M-values for analysis age.mVals <- getM(age.mSetSqFlt) design <- model.matrix(~factor(age.targets$Sample_Group)) # Fit the model for differential variability # specifying the intercept and age as the grouping factor fitvar <- varFit(age.mVals, design = design, coef = c(1,2)) # Summary of differential variability summary(decideTests(fitvar))
## (Intercept) factor(age.targets$Sample_Group)OLD
## -1 0 1325
## 0 11441 393451
## 1 417787 34452topDV <- topVar(fitvar, coef=2) # Top 10 differentially variable CpGs between old vs. newborns topDV
## SampleVar LogVarRatio DiffLevene t P.Value
## cg19078576 1.1128910 3.746586 0.8539180 7.006476 0.0000000006234780
## cg11661000 0.5926226 3.881306 0.8413614 6.945711 0.0000000008176807
## cg07065220 1.0111380 4.181802 0.9204407 6.840327 0.0000000013069867
## cg05995465 1.4478673 -5.524284 -1.3035981 -6.708321 0.0000000023462074
## cg18091046 1.1121511 3.564282 1.0983340 6.679920 0.0000000026599570
## cg05491001 0.9276904 3.869760 0.7118591 6.675892 0.0000000027077013
## cg05542681 1.0287320 3.783637 0.9352814 6.635588 0.0000000032347355
## cg02726803 0.3175570 4.063650 0.6418968 6.607508 0.0000000036608219
## cg08362283 1.0028907 4.783899 0.6970960 6.564472 0.0000000044240941
## cg18160402 0.5624192 3.716228 0.5907985 6.520508 0.0000000053665347
## Adj.P.Value
## cg19078576 0.0001754857
## cg11661000 0.0001754857
## cg07065220 0.0001869984
## cg05995465 0.0001937035
## cg18091046 0.0001937035
## cg05491001 0.0001937035
## cg05542681 0.0001964159
## cg02726803 0.0001964159
## cg08362283 0.0002109939
## cg18160402 0.0002303467Similarly to the differential methylation analysis, is it useful to plot sample-wise beta values for the differentially variable CpGs to ensure the significant results are not driven by artifacts or outliers (Figure 14).
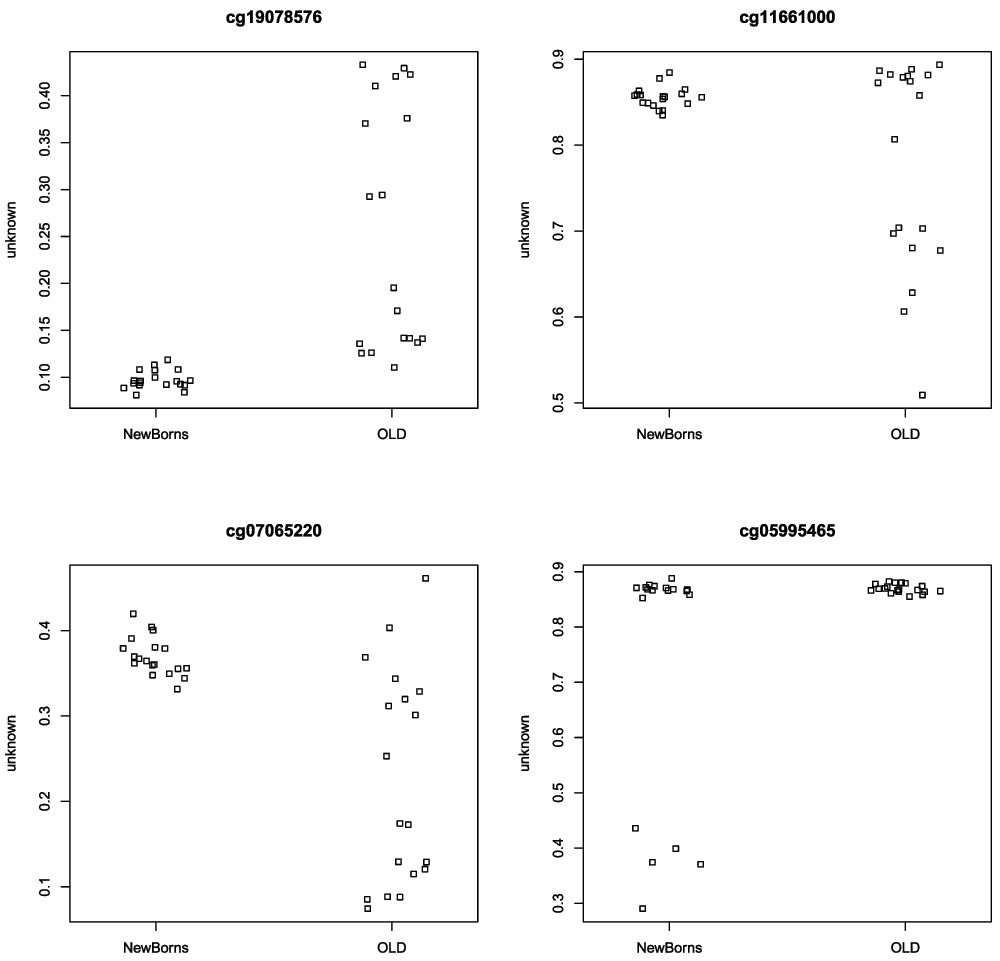
# get beta values for ageing data age.bVals <- getBeta(age.mSetSqFlt) par(mfrow=c(2,2)) sapply(rownames(topDV)[1:4], function(cpg){ plotCpg(age.bVals, cpg=cpg, pheno=age.targets$Sample_Group) })
An example of testing for differential variability when the design matrix does not have an intercept term is detailed in the missMethyl vignette.
As methylation is cell type specific and methylation arrays provide CpG methylation values for a population of cells, biological findings from samples that are comprised of a mixture of cell types, such as blood, can be confounded with cell type composition (Jaffe & Irizarry, 2014). The minfi function estimateCellCounts facilitates the estimation of the level of confounding between phenotype and cell type composition in a set of samples. The function uses a modified version of the method published by Houseman et al. (2012) and the package FlowSorted.Blood.450k, which contains 450k methylation data from sorted blood cells, to estimate the cell type composition of blood samples.
# load sorted blood cell data package library(FlowSorted.Blood.450k) # ensure that the "Slide" column of the rgSet pheno data is numeric # to avoid "estimateCellCounts" error pData(age.rgSet)$Slide <- as.numeric(pData(age.rgSet)$Slide) # estimate cell counts cellCounts <- estimateCellCounts(age.rgSet)
## [estimateCellCounts] Combining user data with reference (flow sorted) data.
## [estimateCellCounts] Normalizing user and reference data together.
## [estimateCellCounts] Picking probes for composition estimation.
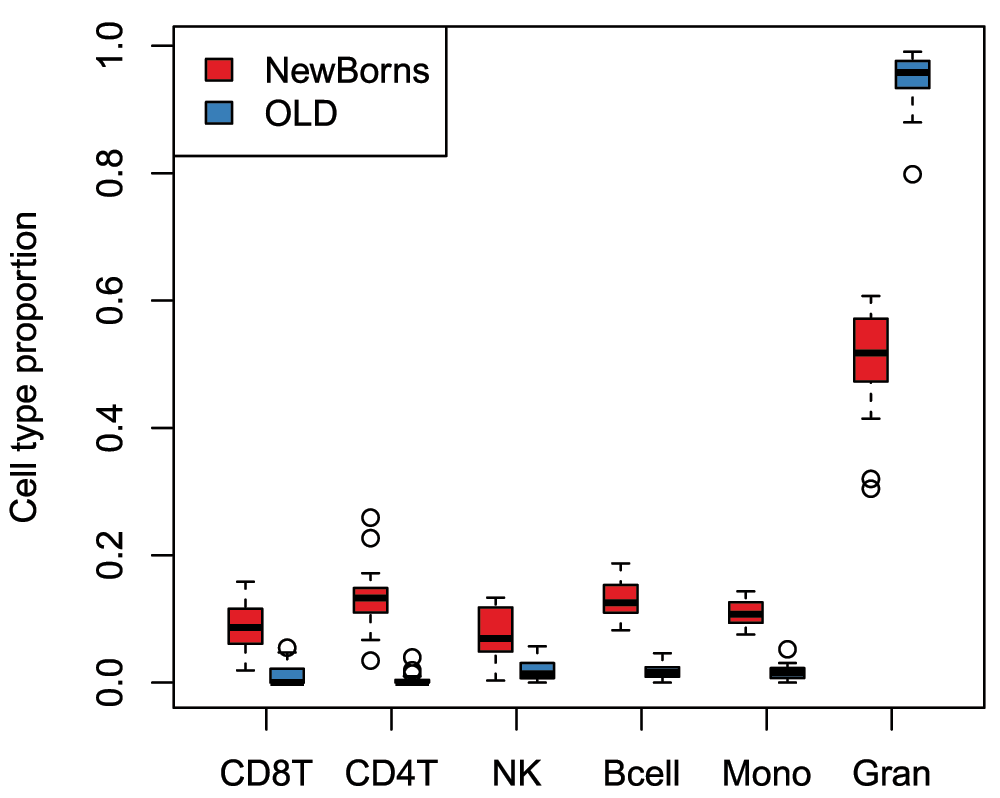
## [estimateCellCounts] Estimating composition.# plot cell type proportions by age par(mfrow=c(1,1)) a = cellCounts[age.targets$Sample_Group == "NewBorns",] b = cellCounts[age.targets$Sample_Group == "OLD",] boxplot(a, at=0:5*3 + 1, xlim=c(0, 18), ylim=range(a, b), xaxt="n", col=age.pal[1], main="", ylab="Cell type proportion") boxplot(b, at=0:5*3 + 2, xaxt="n", add=TRUE, col=age.pal[2]) axis(1, at=0:5*3 + 1.5, labels=colnames(a), tick=TRUE) legend("topleft", legend=c("NewBorns","OLD"), fill=age.pal)
As reported by Jaffe & Irizarry (2014), the plot demonstrates that differences in blood cell type proportions are strongly confounded with age in this dataset (Figure 15). Performing cell composition estimation can alert you to potential issues with confounding when analysing a mixed cell type dataset. Based on the results, some type of adjustment for cell type composition may be considered, although a naive cell type adjustment is not recommended. Jaffe & Irizarry (2014) outline several strategies for dealing with cell type composition issues.
Here we present a commonly used workflow for methylation array analysis based on a series of Bioconductor packages. While we have not included all the possible functions or analysis options that are available for detecting differential methylation, we have demonstrated a common and well used workflow that we regularly use in our own analysis. Specifically, we have not demonstrated more complex types of analyses such as removing unwanted variation in a differential methylation study (Leek et al., 2012; Maksimovic et al., 2015; Teschendorff et al., 2011), block finding (Aryee et al., 2014; Hansen et al., 2011) or A/B compartment prediction (Fortin & Hansen, 2015). Our differential methylation workflow presented here demonstrates how to read in data, perform quality control and filtering, normalisation and differential methylation testing. In addition we demonstrate analysis for differential variability, gene set testing and estimating cell type composition. One important aspect of exploring results of an analysis is visualisation and we also provide an example of generating region-level views of the data.
This workflow uses the following packages available from Bioconductor (version 3.2):
sessionInfo()
## R version 3.2.3 (2015-12-10)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: CentOS release 6.7 (Final)
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] splines grid stats4 parallel stats graphics grDevices
## [8] utils datasets methods base
##
## other attached packages:
## [1] FlowSorted.Blood.450k_1.8.0
## [2] GO.db_3.2.2
## [3] org.Hs.eg.db_3.2.3
## [4] AnnotationDbi_1.32.3
## [5] stringr_1.0.0
## [6] DMRcate_1.6.53
## [7] DMRcatedata_1.6.1
## [8] DSS_2.10.0
## [9] bsseq_1.6.0
## [10] Gviz_1.14.7
## [11] minfiData_0.12.0
## [12] matrixStats_0.50.2
## [13] missMethyl_1.4.0
## [14] RSQLite_1.0.0
## [15] DBI_0.3.1
## [16] RColorBrewer_1.1-2
## [17] IlluminaHumanMethylation450kmanifest_0.4.0
## [18] IlluminaHumanMethylation450kanno.ilmn12.hg19_0.2.1
## [19] minfi_1.16.1
## [20] bumphunter_1.10.0
## [21] locfit_1.5-9.1
## [22] iterators_1.0.8
## [23] foreach_1.4.3
## [24] Biostrings_2.38.4
## [25] XVector_0.10.0
## [26] SummarizedExperiment_1.0.2
## [27] GenomicRanges_1.22.4
## [28] GenomeInfoDb_1.6.3
## [29] IRanges_2.4.8
## [30] S4Vectors_0.8.11
## [31] lattice_0.20-33
## [32] Biobase_2.30.0
## [33] BiocGenerics_0.16.1
## [34] limma_3.26.9
##
## loaded via a namespace (and not attached):
## [1] nlme_3.1-127 bitops_1.0-6
## [3] tools_3.2.3 doRNG_1.6
## [5] nor1mix_1.2-1 rpart_4.1-10
## [7] Hmisc_3.17-3 colorspace_1.2-6
## [9] nnet_7.3-12 methylumi_2.16.0
## [11] gridExtra_2.2.1 base64_1.1
## [13] chron_2.3-47 preprocessCore_1.32.0
## [15] formatR_1.4 pkgmaker_0.22
## [17] rtracklayer_1.30.4 scales_0.4.0
## [19] genefilter_1.52.1 quadprog_1.5-5
## [21] digest_0.6.9 Rsamtools_1.22.0
## [23] foreign_0.8-66 R.utils_2.3.0
## [25] illuminaio_0.12.0 rmarkdown_0.9.6.6
## [27] siggenes_1.44.0 GEOquery_2.36.0
## [29] dichromat_2.0-0 htmltools_0.3.5
## [31] BSgenome_1.38.0 ruv_0.9.6
## [33] gtools_3.5.0 mclust_5.2
## [35] BiocParallel_1.4.3 R.oo_1.20.0
## [37] acepack_1.3-3.3 VariantAnnotation_1.16.4
## [39] RCurl_1.96-0 magrittr_1.5
## [41] Formula_1.2-1 futile.logger_1.4.1
## [43] Matrix_1.2-5 Rcpp_0.12.4
## [45] munsell_0.4.3 R.methodsS3_1.7.1
## [47] stringi_1.0-1 yaml_2.1.13
## [49] MASS_7.3-45 zlibbioc_1.16.0
## [51] plyr_1.8.3 multtest_2.26.0
## [53] GenomicFeatures_1.22.13 annotate_1.48.0
## [55] knitr_1.12.3 beanplot_1.2
## [57] igraph_1.0.1 rngtools_1.2.4
## [59] corpcor_1.6.8 codetools_0.2-14
## [61] biomaRt_2.26.1 mixOmics_5.2.0
## [63] futile.options_1.0.0 XML_3.98-1.4
## [65] evaluate_0.9 biovizBase_1.18.0
## [67] latticeExtra_0.6-28 data.table_1.9.6
## [69] lambda.r_1.1.7 gtable_0.2.0
## [71] reshape_0.8.5 ggplot2_2.1.0
## [73] xtable_1.8-2 survival_2.39-2
## [75] GenomicAlignments_1.6.3 registry_0.3
## [77] ellipse_0.3-8 cluster_2.0.4
## [79] statmod_1.4.24| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
rgSet <- read.450k.exp(targets=targets)
[or the read.metharray.exp function if ... Continue reading Dear David, thank you for your comment. When you read in the methylation data using the targets file specifically in the call:
rgSet <- read.450k.exp(targets=targets)
[or the read.metharray.exp function if using the latest Bioconductor]
this ensures that the order of the samples in the targets file and in the data objects are the same.
rgSet <- read.450k.exp(targets=targets)
[or the read.metharray.exp function if using the latest Bioconductor]
this ensures that the order of the samples in the targets file and in the data objects are the same.