Keywords
whole exome sequencing, mineralocorticoid excess
whole exome sequencing, mineralocorticoid excess
SME: Syndromes of mineralocorticoid excess; AME: Apparent Mineralocorticoid Excess; CAH:17α-hydroxylase: Congenital adrenal hyperplasia due to 17α-hydroxylase deficiency, CAH: 11β-hydroxylase deficiency: Congenital adrenal hyperplasia due to 11β-hydroxylase deficiency; CYP17A1: cytochrome P450, family 17, subfamily A, polypeptide 1; CYP11B1: cytochrome P450, family 11, subfamily B, polypeptide 1, HSD11B2: hydroxysteroid (11β) dehydrogenase 2, ENaC; the epithelial sodium channel subunit genes, WNK1: WNK lysine deficient protein kinase 1, WNK4; WNK lysine deficient protein kinase 1; KLHL3: kelch-like family member 3, CUL3: cullin 3; SPAK: type III secretion system chaperone; MCR/NR3C2: Nuclear Receptor Subfamily 3, Group C, Member 2; WES: Whole exome sequencing.
Syndromes of mineralocorticoid excess (SME) are a group of syndromes characterized by an abnormal activation of the amiloride-sensitive sodium channels in the distal tubules of the kidney resulting in an abnormal salt balance. The symptoms are characteristically an abnormality in salt balance (due to an over activation of the channels mediated through the mineralocorticoid receptor), water retention, hypokalemia, low renin levels and hypertension1–3. Based on the clinical presentation, differential diagnosis would include Liddle syndrome, Geller syndrome, Gordon syndrome, Apparent Mineralocorticoid Excess (AME) and other milder variants such as CAH:17α-hydroxylase and CAH:11β-hydroxylase deficiency. Mutations in various genes including CYP17A1, CYP11B1, HSD11B2, ENaC, WNK1, WNK4, KLHL3, CUL3, SPAK and MCR/NR3C2 may result in clinical features that resemble SME1–3. Due to the large diversity and overlapping clinical features, the accurate diagnosis is highly reliant on the genetic characterization. The need to screen a large number of variants and genes that could cause disease, using conventional approaches such as capillary sequencing is tedious, time consuming and often expensive. Whole exome sequencing (WES) has emerged as an alternative strategy in such clinical settings4,5.
Five siblings of a third degree consanguineous family (Figure 1A) were evaluated at the Department of Nephrology, KMCT Medical College Hospital, Kerala, India. They were aged between 14 and 30 years. All but the youngest of the five siblings were hypertensive. On evaluation, four of the siblings had significant hypokalemia without significant acidosis or alkalosis but with ultrasound evidence of medullary nephrocalcinosis (Figure-1A). The youngest sibling, aged 14 years, did not have hypokalemia, acid base abnormality or renal dysfunction but exhibited overt medullary nephrocalcinosis on ultrasound. Two of the older siblings had concentric left ventricular hypertrophy and renal dysfunction with mild proteinuria. Plasma renin and aldosterone levels were evaluated in the two male siblings and were found to be low (Plasma renin activity 0.14 ng/ml/hr and Plasma Aldosterone 28 pg/ml (sitting upright position) in the elder brother and Plasma renin activity 0.1 ng/ml/hr and Plasma aldosterone 6.5 pg/ml in the younger sibling). Both of them required multiple antihypertensive drugs (Amlodipine 10 mg and Metoprolol Extended Release 50 mg daily) in addition to spironolactone (50 mg daily) for control of blood pressure while the others were well controlled on spironolactone (50 mg daily) alone. There was no past history of similar phenotypes in the parents. A provisional diagnosis of AME was made in view of the clinical picture and inheritance pattern. The 24-hour urinary cortisol to cortisone ratio estimation was not done due to unavailability of the test.
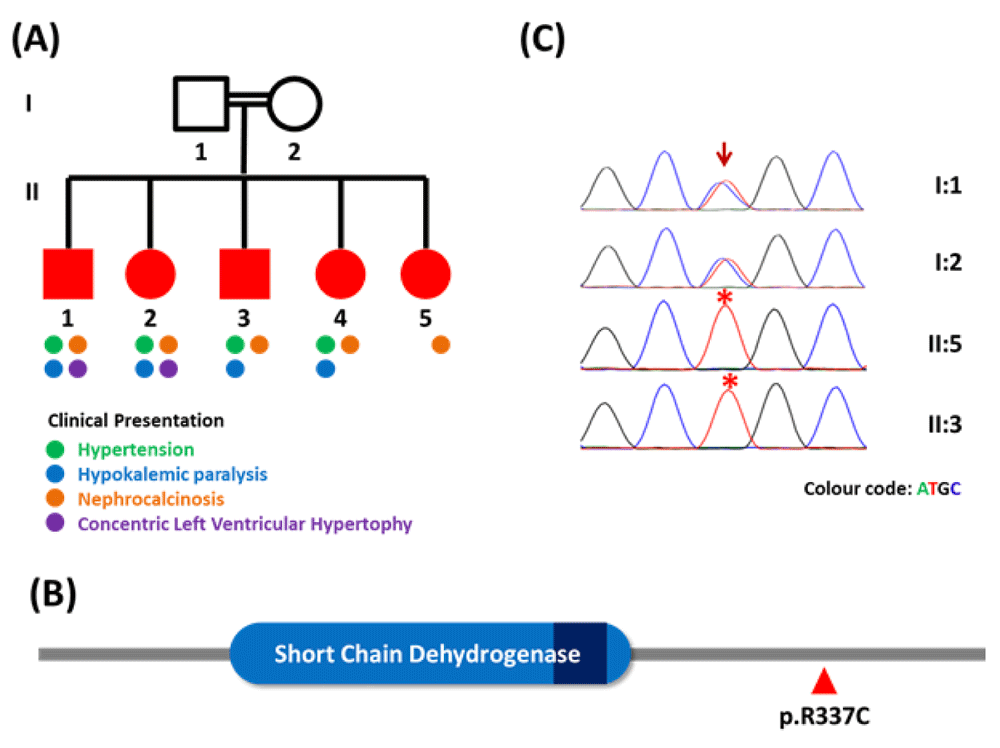
(A) Family pedigree marked with progressive phenotypes. (B) Secondary structure of HSD11B2 marked with major domains and the p.R337C amino acid position. (C) Capillary sequencing chromatogram representation of p.R337C variation in the family; arrow and asterisks marks depict the variation loci and affected individuals respectively.
Whole blood was obtained from the parents and the affected members was obtained after informed consent. 50 ng of the isolated high quality DNA was used to prepare library and exome capture using Nextera Rapid Capture Expanded Exome kit and Sequencing was performed on Illumina Hiseq 2500 sequencer using v3 reagents (Illumina Inc, USA) to generate over 49.48 million paired end reads of 101bp. The variation finding, annotation and prioritisation were performed as previously described5 and revealed the presence of homozygous variation p.R337C in HSD11B2 gene (Figure-1B) annotated to be pathogenic in ClinVar6 and predicted to be deleterious using PROVEAN7. Human cell line studies have demonstrated that p.R337C mutation leads to the low activity of HSD11B2 and causes low-renin hypertension thus resulting in AME8. The variant was further validated using Sanger sequencing of the amplicons, confirming the diagnosis (Figure-1C).
AME is a rare heterogeneous low renin retention SME disorder that manifests with severe juvenile hypertension, hypokalemic alkalosis, low birth weight, failure to thrive, poor growth, and in many cases nephrocalcinosis caused by homozygous and compound heterozygous mutations in the HSD11B2 gene. HSD11B2 oxidises the steroid hormone cortisol to inactive cortisone and mutations in this gene result in a high circulating level of cortisol, and further illegitimate activation of the mineralocorticoid receptor, outcompeting aldosterone and causing activation of the downstream pathways and a clinical presentation of AME symptoms.
In summary, we used WES to characterize and diagnose a family with an extremely rare clinical presentation. p.R337 loci of 11HSDB2 is widely reported to be associated with AME across the world and has been reported in Zoroastrians from India and Iran (Compound heterozygous for p.R337H and Δp.Y338, age of onset from 8 months), in Persian (p.R337C, age of onset from 4 years) and in Japanese (p.R337H and Δp.Y338, age of onset from 2 years) populations9,10. The age of onset for the disease manifestation due to p.R337C mutation has been previously described to be as early as 4 years. Our family, though having the p.R337C mutation, did not present with clinical symptoms in early childhood but exhibits the progressive AME phenotypes with increasing age (Figure 1A) with the older individuals presenting more severe clinical manifestations including renal impairment. Patients with identical homozygous mutations from different families have been described to have varying degrees of severity in clinical and biochemical features4,9. In fact, homozygous mutations in HSD11B2 have been described where the patient has only mild low renin hypertension without other features of AME10. Early diagnosis and prompt institution of salt restriction and spironolactone in these patients can prevent secondary organ damage. Our report also serves to highlight the utility of WES as a tool for diagnosing rare genetic diseases even where biochemical characterization is unavailable.
Written informed consent for publication of these data were obtained from the patients.
The raw whole exome sequence are available at the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra), accession number SRR3546815.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)