Keywords
structural chromosomal aberrations, recurrent breakpoint genes, molecular characterization, cancer genome, copy number aberration profile, computational method
This article is included in the Bioconductor gateway.
This article is included in the RPackage gateway.
structural chromosomal aberrations, recurrent breakpoint genes, molecular characterization, cancer genome, copy number aberration profile, computational method
In this version we provide a much more extensive description of the underlying statistics for the detection of recurrent breakpoint events on genomic location- and gene-level. In addition, we rephrased a few sentences.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Tumor development is driven by irreversible somatic genomic aberrations such as single nucleotide variants (SNVs) and chromosomal aberrations including numerical as well as structural changes1,2. Genome-wide somatic DNA copy number aberrations (CNA) profiling is a widely established approach to characterize chromosomal aberrations in cancer genomes. At present, application of computational methods has mainly been focused on the analysis of numerical aberrations of chromosomal segments. Evidence is emerging that genes affected by structural chromosomal aberrations, i.e. genes affected by chromosomal breaks, represent a biologically and clinically relevant class of mutations in many cancer types including solid tumors3–6. Importantly, the actual locations of chromosomal CNA-associated breakpoints, which are the points of copy number level shift in somatic CNA profiles, indicate underlying chromosomal breaks and thereby genomic locations affected by somatic structural aberrations5–12. Hence, the wide availability of large series of high-resolution DNA copy number data by for instance array-Comparative Genomic Hybridization (CGH) or by (low-pass) whole genome sequencing (WGS) approaches enables to systematically search for regions and genes that are affected by CNA-associated structural chromosomal changes. Computational methods determining numerical CNAs, consequently, also yield CNA-associated breakpoint locations. However, it is not trivial to identify genes that are recurrently affected by CNA-associated chromosomal breakpoints across (large) series of cancer samples since this methodology also requires dedicated computational methods including comprehensive statistical evaluation.
We here provide a computational method, ‘GeneBreak’, that identifies chromosomal breakpoint locations using DNA copy number profiles. A tailored annotation approach maps breakpoint locations to genes for each individual profile. Moreover, dedicated comprehensive cohort-based statistical analysis including correction for covariates that influence the probability to be a breakpoint gene and multiple testing pinpoints genes that are non-randomly and recurrently affected by chromosomal breaks across multiple tumor samples5. ‘GeneBreak’ is implemented in R (www.cran.r-project.org) and is available from Bioconductor (www.bioconductor.org/packages/release/bioc/html/GeneBreak.html). The Bioconductor vignette describes a detailed example workflow of CNA data obtained by analysis of 200 array-CGH samples. A schematic overview of computational methods is depicted in Figure 1.
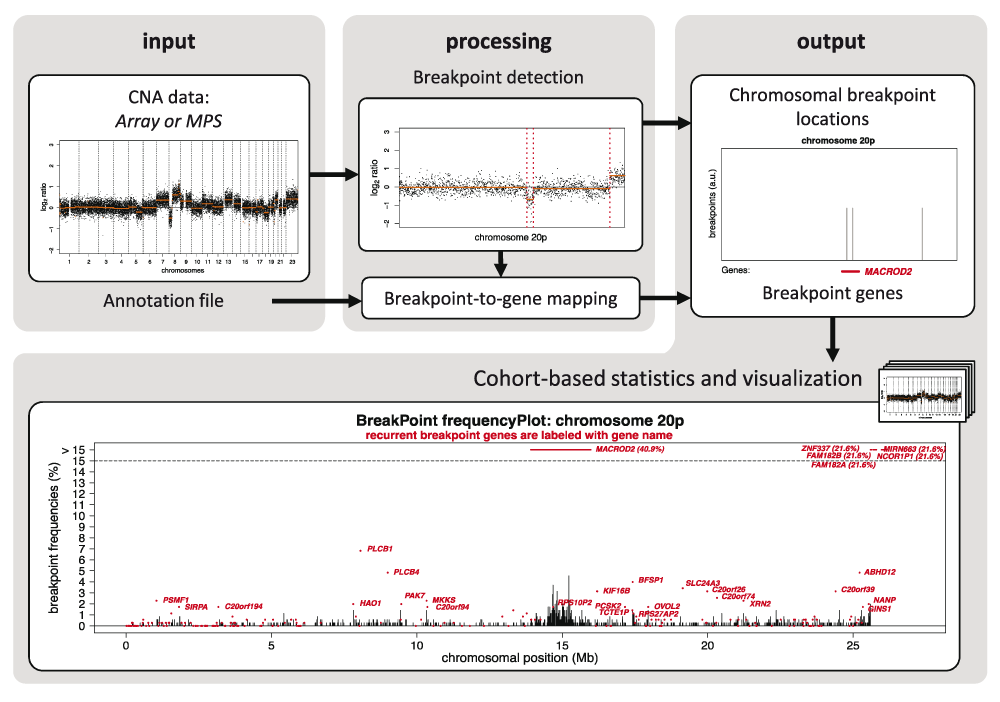
GeneBreak’ requires already segmented DNA copy number data from array-CGH or WGS approaches. The first step involves detection of breakpoint locations. Next, breakpoint locations will be mapped to gene annotations in order to identify genes affected by DNA breakpoints. The final step performs comprehensive cohort-based statistical analyses including correction for multiple testing to reveal both recurrent breakpoint locations and breakpoint genes. The breakpoint frequencies can be visualized with a built-in plot function. This example visualizes the breakpoint locations (vertical black bars) and breakpoint genes (horizontal red bars) on the p-arm of chromosome 20 identified in a cohort of 352 advanced colorectal cancers. The genes labeled with a name are statistically significant recurrent breakpoint genes (FDR<0.1).
The breakpoint detection method we provide is amenable for data from any DNA copy number discovery platform, e.g. array-CGH and (low-pass) WGS. For optimal results, ‘GeneBreak’ takes DNA copy number data that are pre-processed by the R-package ‘CGHcall’13 or ‘QDNAseq’14, both based on the Circular Binary Segmentation algorithm15, as input. Alternatively, segmented values (log2-ratios) from a different copy number detection algorithm can be used. In addition, it is recommended to provide discrete DNA copy number states (e.g. loss, neutral, gain) that can be used for breakpoint selection. Bioconductor vignette and manual describe commands and workflows in detail (See Supplementary material).
Breakpoints are defined by the chromosomal locations that separate the contiguous DNA copy number segments pinpointed by a segmentation algorithm. ‘GeneBreak’ identifies chromosomal breakpoint locations for each individual DNA copy number profile. Instead of taking all detected breakpoints, users may want to define more precisely what breakpoints to take into account, based on the two flanking DNA copy number segment characteristics. One of the following three selection options can be applied. A) Copy number-deviation: this selects breakpoints where the shift in log2-ratio between two consecutive DNA copy number segments exceeds the user-defined threshold; B) CNA-associated breakpoints: this selects all breakpoints between consecutive DNA copy number segments, except for breakpoints flanked by two copy number neutral segments; C) CNA-breakpoints: this selects only those breakpoints flanked by segments with dissimilar discrete DNA copy number states.
Breakpoints are defined by the genomic start position of the copy number segments. DNA copy number profiling data is typically granular due to the distance between microarray probes or bin size of WGS copy number data. This means that the genomic location of a breakpoint is not detected at nucleotide resolution but represents a chromosomal interval with a size that is determined by microarray probe density or WGS bin size.
For identification of genes affected by chromosomal breakpoints the built-in gene annotations can be used. Alternatively, a user-defined gene annotation file can be provided (see Bioconductor vignette and manual for further details). The implemented mapping approach identifies genes that are associated with one or multiple chromosomal breakpoint intervals.
Identification of statistically recurrent breakpoint events across all samples can be performed on both chromosomal location- and gene-level. As features, i.e. microarray probes or bins of WGS copy number data, are (nearly) equally distributed over the genome, we assume that the null- probability for breakpoint occurrence is equal for all individual candidate breakpoints (features). It differs per sample, though, and equals ps = Ns/N, where N is the number of probes, and Ns the total number of breakpoint for samples. The test statistic is Tp is the total number of breakpoints for probe p across all samples. Then, under the null-hypothesis, Tp is simply a sum of independent Bernoulli (ps) random variables, the null-distribution of which is the same for all probes. It is quickly computed by using probability generating functions, giving also the p-values for any observed value of Tp.
The probe-based statistical analysis uses Benjamini-Hochberg false discovery rate (FDR) correction for multiple testing. For the intended use at gene level, a more advanced statistical null-model is required. For the gene level, the null-probability for a breakpoint to occur within an individual gene, depends on 1) the length of the gene, 2) the number of gene-associated features and 3) the number of breakpoints in the entire tumor profile for the specific sample. Therefore, at gene-level, we apply a linear regression-based correction for covariates. These regression-estimates are then used as gene- and sample-specific breakpoint null-probabilities (pg,s). The test statistic remains the same, and so does the null-distribution computation, although it has to be repeated for each gene now. Finally, the Gilbert FDR correction that accounts for discreteness in the null-distribution16 is applied in this analysis to determine significance of recurrent breakpoint genes. Commands and example workflow can be found in Bioconductor vignette and manual.
We applied our method to 352 high-resolution array-CGH samples from a series of advanced colorectal cancers17 following CNA detection using ‘CGHcall’13. Array-CGH data are available in the Gene Expression Omnibus database under accession number GSE63216 (www.ncbi.nlm.nih.gov/projects/geo/). We selected for the CNA-associated breakpoints (setting: ‘CNA-associated’), used gene annotations from ensembl (human genome NCBI build36/hg18, release 54) and applied the dedicated Benjamini-Hochberg-type FDR correction (setting: ‘Gilbert’), for recurrent breakpoint gene identification. A total of 748 genes appeared to be recurrently affected by chromosomal breaks (FDR<0.1)5. Breakpoint frequencies of chromosome 20p are visualized with the built-in plot function (Figure 1; see Bioconductor vignette and manual for further details about this function). Interestingly, patient stratification based on recurrent gene breakpoints and well-known point mutations by propagation to the predefined STRING human protein interaction network revealed one CRC subtype with very poor prognosis, which supported clinical relevance of this class of somatic aberrations in advanced colorectal cancers5.
Genome instability including numerical and structural somatic chromosomal aberrations is a hallmark of cancer. Several tools are available that focus on detection of numerical aberrations of large chromosome segments. The R-package ‘GeneBreak’ extracts additional information from CNA data. ‘GeneBreak’ provides an easy-to-use algorithm, which handles identification of genomic breakpoint locations, mapping of breakpoints to genes and includes a comprehensive statistical approach to reveal recurrent breakpoint genes from series of tumor samples. Therefore, ‘GeneBreak’ can be applied to detect CNA-associated chromosomal breaks in individual tumor samples and facilitates detection of recurrent breakpoint genes across multiple tumor samples.
Publicly available copy number data used for the use case is deposited at Gene Expression Omnibus database under accession number GSE63216 (https://protect-eu.mimecast.com/s/6LQhBmNGvCG).
Software available from: C www.bioconductor.org/packages/release/bioc/html/GeneBreak.html and https://protect-eu.mimecast.com/s/aLGhBqmpgF2
Latest source code: https://github.com/F1000Research/GeneBreak/releases/tag/v1.0
Archived source code as at the time of publication: F1000Research/Genebreak, doi: 10.5281/zenodo.15393718
License: GPL 2
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)