Keywords
RNA-seq, transcriptome, Computational genomics, chickpea, Cicer arietinum, re-validation, Intermediate assembly data, Big Data, Bioproject
RNA-seq, transcriptome, Computational genomics, chickpea, Cicer arietinum, re-validation, Intermediate assembly data, Big Data, Bioproject
The lack of reproducibility of results in biology is a contentious subject1,2. In computational studies, the exact replication of the output of most computer programs is difficult as most non-trivial algorithms use heuristics. The problem is compounded by recent technological advances generating "Big Data" involving multiple programs and pipelines3,4. However, inferences based on these results should not be subject to the same, or ideally any, unpredictability. The availability of software used at each stage and the intermediate data generated is key in enabling debugging and tracking the veracity of results by subsequent researchers5.
Chickpea (Cicer arietinum L.) is an important pulse crop having numerous nutritional and health benefits6. Several online resources exist for chickpea genomes and transcriptomes (http://www.cicer.info/databases.php, http://www.nipgr.res.in/ctdb.html, http://gigadb.org/dataset/100076, http://nipgr.res.in/CGAP/7). Interestingly, the 68th United Nations General Assembly has declared 2016 as the International Year of Pulses (IYP).
The RNA-seq8,9 derived transcriptome of chickpea has also been sequenced10. In contrast to other traditional methods like RNA:DNA hybridization11 and short sequence-based approaches12, RNA-seq detects transcripts with very low expression levels. YeATS is a work-flow for analyzing RNA-seq data13, and was used to detect a second homolog of a polyphenol oxidase gene and ~130 genes in the large gallate 1-β- glucosyltransferase in walnut14. YeATS analysis of RNA-seq data from 20 different tissues of walnut in California unravelled detailed, tissue-specific information of ~400 transcripts encoded by a large family of resistance (R) genes and elucidated the biodiversity and possible plant–microbe interactions15.
In the current work, errors arising from the assembly step as identified by YeATS are shown to have a bearing on the transcript quantification. The chickpea transcriptome (http://www.nipgr.res.in/ctdb.html10) and its quantification in different tissues provided by an online interface is analyzed. This demonstrates that transcripts which are tagged with high counts predominantly encode multiple genes. While some studies provide the assembled sequences16,17, the author could not find the relevant data, even after personal communication18. It is also proposed that the availability of intermediate assembly sequences be made mandatory, in line with the recent initiative Global Open Data (GODAN: http://www.godan.info/).
The transcriptome of the Cicer arietinum (transHybrid.fasta, ICC4958; Desi chickpea) “represents optimized de novo hybrid assembly of 454 and short-read sequence data. About 2 million 454 reads were assembled using Newbler v2.3 followed by hybrid assembly with 53 409 transcripts generated by optimized short-read data assembly reported previously10 using TGICL program” (http://www.nipgr.res.in/ctdb.html10). Quantification of transcripts in different tissues is provided by an online interface. The chickpea genome (Cicer_arietinum_GA_v1.0.fa) is obtained from http://gigadb.org/dataset/10007619.
YeATS13 analyzed the post assembly transcripts, and first excluded transcripts that did not align to the genome. A BLAST database of protein peptides (plantpep.fasta:1M sequences) using ~30 organisms (list.plants) from the Ensembl genome was created20. The three longest ORFs were obtained using the ‘getorf’ utility in the EMBOSS suite21. These ORFs are BLAST’ed22 to the 'plantpep.fasta’. We identify three classes of errors. Type I error occurs when a single transcript has multiple ORFs with significant matches to different genes. In a Type II error a single gene is broken into two separate transcripts. For a type III error, a single transcript has multiple ORFs, but they all map to the same gene. Multiple sequence alignment was done using MAFFT (v7.123b)23, and sequence alignment figures were generated using the ENDscript server24.
The chickpea transcriptome (transHybrid.fasta:n=3476010) was first mapped to the chickpea genome (Cicer_arietinum_GA_v1.0.fa) obtained from http://gigadb.org/dataset/100076. There were 60 unmapped transcripts, some of which are mitochondrial transcripts (list.mito in Dataset1), some are metagenomic contamination (list.meta in Dataset1), and the rest have no match in the complete BLAST ‘nt’ database (list.nomatchinNT in Dataset1). The metagenomic transcripts are removed from further processing.
Subsequently, each transcript was split into three ORFs (list.3ORFS:n=104K in Dataset1), each of which was BLAST’ed22 to a subset of plant proteins created from the Ensembl20 database (see Methods).
There are ~1300 transcripts encoding more than one significant peptide (see list.duplicate in Dataset1). The top five highly expressed transcripts (HET) from five tissues - flower bud (FB), mature leaf (ML), root (RT), shoot (SH), young plant (YP) - were obtained from http://www.nipgr.res.in/ctdb.html (tissues.txt in Dataset1). The number of transcripts encoding multiple genes, as found from ’list.duplicate’, were are follows - FB:4, ML:5, RT:3, SH:4 YP:5, indicating an over-representation of merged transcripts in HET.
The top five HET in the root, and the genes encoded by them are listed in Table 1. The ORFs of TC00004 encoded in the reverse direction map to a partial retinoblastoma-binding-like protein (~500 out of 549 amino acids) and the complete senescence-associated protein (157 amino acids) (Figure 1). TC00004 is computed to have highly transcribed (ranked in the top five) in all five tissues studied here (tissues.txt in Dataset1). The top ranking transcript, TC00002, has two ORFs - ORF.11 and ORF.38 (TC00002.orf in Dataset1) - which align to an ATP synthase subunit beta (327 aa) and senescence-associated protein (615 aa), respectively. This transcript is highly fragmented and encodes on both strands in an overlapping manner.
TrLen: length of transcript, PLen: length of the full protein, OLen: length of ORF, RPKM: Reads Per Kilobase of transcript per Million mapped reads. Three out of the five transcripts have Type I errors, where two different transcripts are merged.
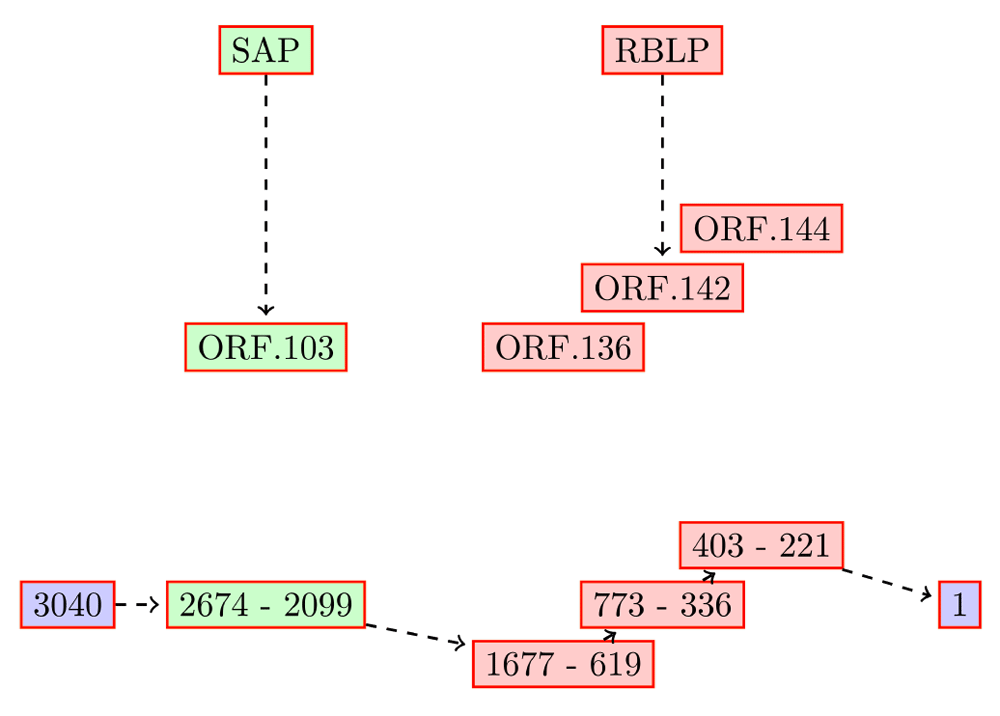
The RBLP ORFs are fragmented, and combine to ~500 aa. TC00004 is computed to be highly transcribed (ranked in the top five) in all five tissues studied here.
Since the merged transcripts are proximally located in the genome, it is possible that these loci are under the same transcriptional control and the expression counts are correct. However, the over-representation of such merged transcripts in HET suggests that there might be some errors in counting.
There are transcripts that encode fragmented ORFs which map to the same protein. YeATS has a merging algorithm that identifies overlapping amino acid sequences in transcripts. For example, TC13991 (35 aa) and TC23009 (110 aa) have an overlapping amino acid sequence "ASNGGRVHC", and both map to a ribulose bisphosphate carboxylase small chain (RBCSC, 180 aa) family protein (Figure 2). TC13991 has a count of 17.90, while TC23009 has a count of 1403.8. The large difference indicates an erroneous normalization algorithm, since there is only one expressed transcript of this gene (the ORF of TC13991 has no other significant match among other transcripts), although there are other genomic variants. TC23009 is ranked fifth among HET in the shoot, while all better ranked transcripts are transcripts having Type I errors. Furthermore, there is a missing 35 aa stretch in the C-terminal peptide "IGFDNVRQVQCISFIAHTPKEF", which has no match in the transcripts. A similar scenario with fragmented transcripts and a missing fragment was detected by YeATS in a polyphenol oxidase gene in walnut14.

Also, their counts are significantly different - TC13991 has a count of 17.90, while TC23009 has a count of 1403.8, indicating an erroneous normalization algorithm. The ORF of TC13991 does not have other significant matches among other transcripts. Furthermore, the C-terminal peptide "IGFDNVRQVQCISFIAHTPKEF" has no matches in the transcripts.
There are ~3000 transcripts which encode more than one ORFs mapping to the same peptide (list.splitORF in Dataset1). TC01688 is one such transcript having ORF.70 and ORF.89 (see TC01688.orf in Dataset1) mapping to an aspartyl protease family protein (TAIRid:AT1G05840.1) with BLAST bitscores 250 and 285, respectively. Merging ORF.70 and ORF.89 (inserting ‘ZZZ’) results in an increased BLAST bitscore of 507 (Figure 3). This should have minimal effects on the counts, unless Type I or Type II also occur simultaneously.
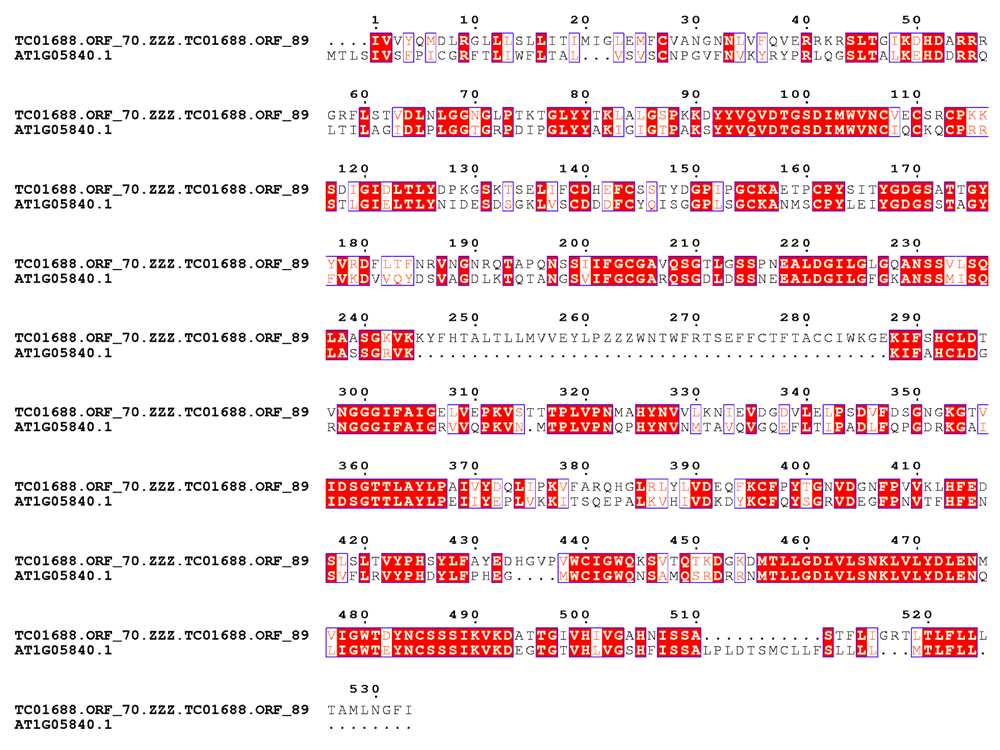
The BLAST bitscore of the merged ORF increases to 507. Errors like this should have minimal effects on the counts, unless Type I or Type II also occur simultaneously.
In the current work, assembler errors have been categorized into three types. These errors have been analyzed for the chickpea transcriptome sequence, and anomalies in the quantification have been detected. The availability of assembled transcriptome sequence has enabled such analysis. It is proposed that sequences of assembled transcriptomes be linked to the Bioproject. Such initiatives have been adopted in the Global Open Data (http://www.godan.info/pages/statement-purpose) to “to make agricultural and nutritionally relevant data available, accessible, and usable for unrestricted use worldwide”.
F1000Research: Dataset 1. Raw data for YeATS on chickpea transcriptome, 10.5256/f1000research.9667.d13681625
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)