Keywords
acute myocardial infarction, stem cell mobilization, preconditioning, SDF-1/CXCR4 axis, MMP-2
acute myocardial infarction, stem cell mobilization, preconditioning, SDF-1/CXCR4 axis, MMP-2
The new version of the manuscript describes in more detail the study design, particularly with regard to the analysed time window.
In the third version we have included a couple of sentences in the discussion and limitation section taking into consideration the comments from the reviewer.
See the authors' detailed response to the review by Amadou K.S. Camara
See the authors' detailed response to the review by Zhengyuan Xia and Michael Irwin
Heart regeneration after ischemic insult is still a matter of debate in spite of extensive research conducted in this field. One of the endogenous cardiac repair mechanisms is the mobilization of regenerative cells derived from bone marrow (BM), followed by migration and homing of the cells in the ischemic myocardial tissue1. Several factors have been identified that play a role in the mobilization of BM-origin stem and progenitor cells, and assist in migration and homing, such as chemotactic factors, complement fractions, cytokines, microRNAs or microvesicles. Among these substances, the axis of the stromal-derived factor-1 alpha [SDF-1α; chemokine receptor 12 (CXCL12)] and chemokine receptor type 4 (CXCR4) exerts the strongest chemoattractant stimulus for migration and homing of cells in the BM and tumors, but also in ischemic tissues, such in case of myocardial ischemia or ischemic stroke2. The local upregulation of SDF-1α attracts the cells covered with CXCR4 receptors towards the SDF-1α gradient, facilitating the migration of the cells into the target organ tissues.
Exploiting the beneficial effect of the SDF-1α/CXCR4 axis in cardiac repair has been performed by repeated injections of granulocyte-colony stimulating factor (G-CSF) applied in patients, which aims to release the stem and premature cells from BM and activate the cellular CXCR4 expression of the reparative cells by interrupting the BM-SDF-1α/CXCR4 axis3. However, despite the enhanced cell release and migration, and stimulation of the endogenous cardiac progenitor cells, the efficacy of clinical cardiac cell-based therapy in patients with recent acute myocardial infarction (AMI) led to ambiguous results4, especially if the regenerative cell therapy was performed very early after ischemic injury5.
Among several mechanisms explaining the cardiac regenerative processes, secretion of distinct chemokines, cytokines, and growth factors may play an important role in the course of events of myocardial infarction2. Tang et al demonstrated upregulated SDF-1α expression in infarcted mouse myocardial tissue after implantation of mesenchymal stem cells simulated by vascular endothelial growth factor (VEGF). This led to increased mobilisation of BM-derived stem cells6. Also, the upregulation of pro-inflammatory cytokines, such tumor necrosis factor (TNF)α7 and interleukin (IL)-88, might initiate processes triggering increased cell trafficking, since myocardial infarction is associated with the inflammatory response8. In contrast, matrix metalloprotease (MMP)-2 cytokine is known to be the inhibitor of SDF-1α, implicating its inactivation9. Another mechanistic process is cardioprotection, induced by either ischemic pre-, post- or remote conditioning, or by release of numerous cardioprotective substances, but their clinical importance is still doubtful. Ischemic preconditioning (IP) has been shown to exhibit cardioprotective mechanisms, and stimulates the recruitment and homing of progenitor cells toward ischemic myocardium in the early phases of cardioprotection in several animal models7. IP-induced cardioprotection possesses bi-phasic effect by initiation of protective mechanisms in the early phase appearing within the first 3 hours following myocardian insult10 and in the late phase, which re-appear 24 hours and lasts for 72 hours11. In our study, we focused on the late window of cardioprotection in relation with stem cell mobilisation and cytokine releases.
In our present experiment, we have investigated the mobilization of BM-origin CD34+ cells 3 days after reperfused AMI in relation to the SDF-1α/CXCR4 axis in a clinically relevant in vivo pig model. We conducted the experiments in domestic female pigs, since the female pigs tolerate the ischemic burden better regarding mortality as compared to male pigs12.
We measured the release of several cytokines, such as MMP-2, VEGF, fibroblast growth factor (FGF)-2, IL-8 and TNFα, to investigate and explain the possible mechanisms behind insufficient cardiac repair in the first days post-AMI. In addition, the current study explored possible additional benefits elicited by IP that involve release of cytokines, CD34+ cells, and MMP-2 expression.
Randomly selected female domestic pigs (n=21; weight, 30–35kg) underwent percutaneous coronary intervention (PCI) under general anaesthesia in order to perform either ischemic preconditioning (group IP; n=6) or non-conditioned AMI (group control; n=12), with a block 1:2 randomisation, due to expected higher mortality in the control group. Animals in both groups underwent 90min percutaneous balloon occlusion of the left anterior descending (LAD) coronary artery at the origin of the first diagonal branch following reperfusion (balloon deflation). IP was initiated prior to 90min LAD occlusion by 3×5min repetitive cycles of artery re-occlusion and reperfusion. One pig in the IP and two animals in the control group died during the AMI intervention, all remaining animals (n=6 in IP and n=12 in control group) survived for 1 month after the experimental procedure (Figure 1).
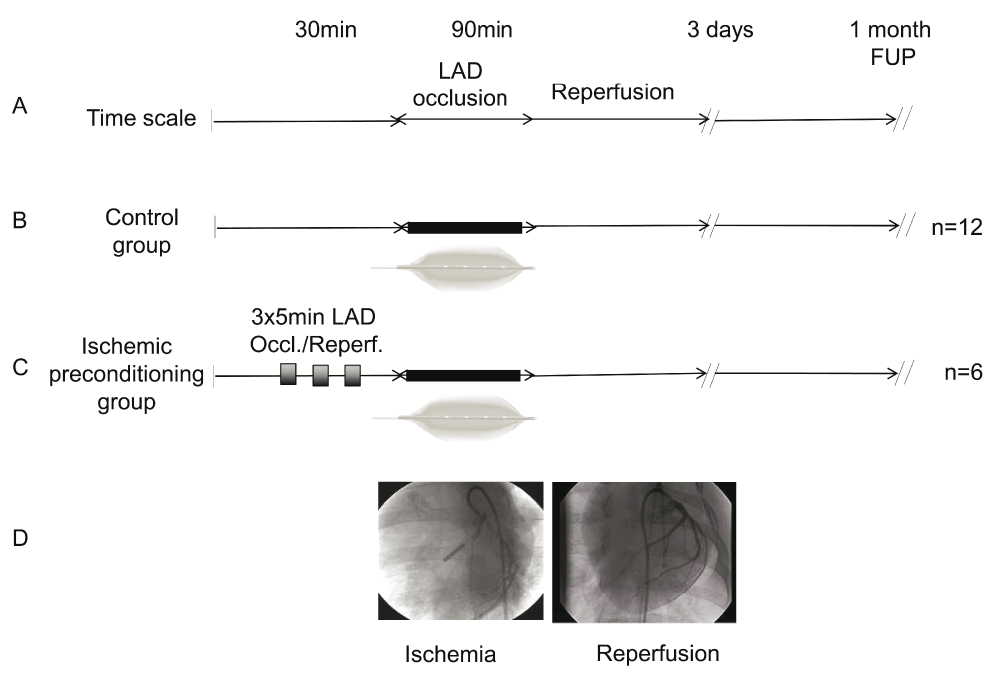
(A) Time scale: 30min anaesthesia, followed by 90min occlusion of the mid left anterior descending coronary artery (LAD), followed by reperfusion. Follow-up (FUP) times 3 days and 1 month. (B) Control group (n=12). (C) Ischemic preconditioning group (n=6) was induced by 3×5min cycles of ischemia/reperfusion (balloon inflation/deflation) prior to 90min balloon occlusion of the LAD. (D) Angiographic pictures of the balloon occlusion of the LAD (left, left anterior oblique acquisition at 45°) and control angiography after restoration of the reperfusion (right, anteroposterior view).
All procedures were performed with the approval of the local Experimental Animal Care Committee (EK SOI/31/26-11/2014) of the University of Kaposvar, Hungary, conforming to the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication No. 85–23, revised 1996). All animal experiments were conducted at the institute of diagnostic imaging and radiation oncology, University of Kaposvar.
Female domestic pigs received 12mg/kg ketamine, 1mg/kg xylazine and 0.04mg/kg atropine as anaesthesia. The anaesthesia was deepened via mask maintaining 1.5–2.5 vol % isofluran, 1.6–1.8 vol % O2 and 0.5 vol % N2O. In total, 200IU/kg of heparin was administrated via the right femoral artery, and selective angiography of LAD arteries was performed prior to induction of myocardial ischemia (MI). MI was induced by 90min balloon occlusion (3.0mm ø, 15mm length, 5atm; Maverick, Boston Scientific, MA, USA) at the mid-part of the LAD artery following balloon deflation. The % O2 saturation, blood pressure and electrocardiogram were continuously measured during the intervention.
Blood samples were collected from the femoral vein for the detection of biological markers. Samples were centrifuged at 2000×g for 10min, and the plasma and serum samples were stored at -20°C until the analysis was performed. For fluorescent activated cell sorting (FACS) analysis, whole blood was collected into EDTA-treated tubes (BD Vacutainer®; Becton, Dickinson and Company, New Jersey, USA) at baseline, 3 days post MI and 1 month follow-up (FUP). All blood samples were processed within 6h.
Plasma level of stromal cell-derived factor-1 (porcine SDF-1α ELISA Kit; Neoscientific, Germany), chemokine (C-X-C motif) receptor 4 (pig CXCR4 ELISA Kit; Abbexa, UK), 72kDa isoform of matrix metalloproteinase-2 protein (porcine MMP-2 ELISA Kit; MyBioSource, CA, USA), fibroblast growth factor-2 (porcine FGF-2 ELISA Kit; Neoscientific, Germany), and vascular endothelial growth factor (porcine VEGF ELISA Kit; Neoscientific, Germany) were detected using commercial ELISA kits, according to the manufacturer’s instructions. Tumor necrosis factor alpha (Porcine TNFα Quantikine ELISA Kit; R&D Systems, MN, USA), and interleukin-8 (pig IL-8 ELISA Kit; Abcam, UK) were detected from serum, according to the manufacturer’s instructions.
Absorbance readings at wavelength 450nm were performed on the automated plate reader VIKTOR3 (Perkin Elmer, MA, USA), and the resulting values were determined by interpolation from a standard curve. Measurements were performed in duplicates. Plasma or serum levels of markers were measured at baseline, 3d post MI and at 1 month FUP.
FACS analysis of whole blood samples was performed at baseline, 3d post MI and at 1 month FUP in order to address the kinetics of mobilized CD34+ cells in vivo. EDTA-treated venous blood samples (100μl) were labelled with PE-DY647-conjugated CD34+ antibody (monoclonal antibody; host/isotype: mouse/IgG1; cat# MA1-19770; Thermo Fisher Scientific, Waltham, MA, USA) or the corresponding isotype control (PE-conjugated mouse IgG1; cat# MA1-10415; Thermo Fisher Scientific, Waltham, MA) for 20min at room temperature. Anti-human CD34+ antibody was utilized due to lack of commercially available porcine-specific CD34+ marker (dilution: 5μl antibody/100μl whole porcine blood). Subsequently, erythrocyte cell lysis was performed, according to the manufacturer’s protocol, using Dako-Uti LyseTM (Dako, Agilent Technologies, Santa Clara, CA, USA) following fixation with PBS containing 1% paraformaldehyde. FACS analysis was performed on CyFlow® ML/space flow cytometer (Sysmex Partec, Görlitz, Germany) with acquired 100.000 events within the gated region of mononuclear cells of forward versus side scatter. Absolute counts of CD34+ cells were obtained by multiplying the ratio of the CD34+ cells obtained in the flow cytometry analysis and absolute count of leucocytes per 1μl of blood.
Human adult cardiac myocytes (HACMs) were isolated from the left ventricular tissue obtained from the hearts of patients undergoing heart transplantation. Mechanical dissociation of the tissue and separation of the cardiomyocytes from fibroblasts detached to Petri-dish surface was performed, as described previously13. All tissue donors gave their informed written consent to the study. The study was approved by the local ethical committee (Medical University of Vienna, Austria; EK 151/2008) and complies with the Declaration of Helsinki.
Human cord blood CD34 positive cells (CD34+ cells) were purchased from StemCell Technologies Company (Grenoble, France). The cells were used in in vitro cell migration assay to assess their migratory capacity toward HACMs. Since porcine CD34+ cells are not commercially available, we used human cardiomyocytes and human cord blood CD34+ cells.
Migration of CD34+ cells was monitored by commercially available Roche xCELLigence System (Acea Bioscience, CA, USA), according to the manufacturer’s instructions. Briefly, 160μl suspension of HACM cells (conc. 10.000 cells/well) was resuspended in M199 cardiac cell culture media (Sigma-Aldrich, Vienna, Austria) containing 20% FBS and 1% Pen/Strep solution (Gibco™, Thermo Fischer Scientific, MA, USA). Cell suspension was transferred to the lower chamber of the CIM-Plate with integrated gold microelectrode sensors (Figure 2).
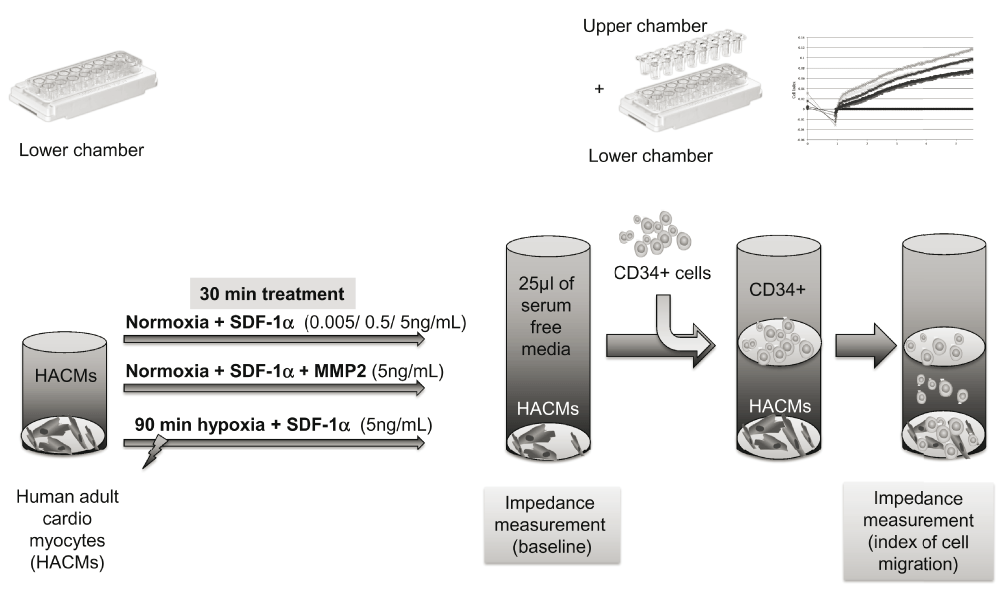
Human adult cardiac myocytes (HACMs) were incubated in the lower chamber. After 30min incubation with different treatments: either adding stromal-derived factor-1-alpha (SDF-1α) in increasing concentrations, or adding the highest concentration of SDF-1α and matrix-metalloprotease-2 (MMP-2), or the cells were kept under hypoxia for 90min, followed by the addition of SDF-1α. Lower and upper chamber were then combined, and after adding serum-free media to the upper chamber, baseline impedance measurements were performed, followed by adding CD34+ cells. Impedance measurements were performed to quantify the CD34+ cell migration towards HAMCs.
HACMs were incubated under normoxic conditions with increasing doses (0.005, 0.5 and 5.0ng/mL) of SDF-1α (Sigma-Aldrich, Vienna, Austria) to evaluate the maximal SDF-1α chemoattractant effect on CD34+ cells towards HCAMs.
In order to analyse the effect of MMP-2 and hypoxia on the mobilisation of CD34+ cells toward HCAMs, HCAMs were incubated with SDF-1α (with the elaborated maximal effect of 5.0ng/mL) either with co-incubation with MMP-2 (5.0ng/mL; Sigma-Aldrich, Vienna, Austria) under normoxia, or under 90min hypoxic conditions.
Hypoxia (90min, 37°C, 1% O2) was induced in HCAM cell culture on the CIM-Plate by sealing the cell culture plate in an airtight plastic bag (Microbiology Anaerocult® IS Bag; Merck Millipore, Vienna, Austria) containing a dry anaerobic indicator strip.
In total, 25μl serum-free medium (M199 containing 0.1% FBS) was added to the upper chamber 30min after treatments with the various substances, and the chambers were combined for background measurements. Subsequently, CD34+ cells (100.000 cells/well) were transferred to the upper chamber of the CIM-Plate with polyethylene terephthalate membrane (PET) with 8μm pore diameter and measurements were repeated. Migrated cells translocated through the PET-membrane and changed the impedance signal captured by sensors in the lower chamber. The background was subtracted from all results and each experiment was repeated three times (Figure 2).
Continuous parameters were expressed as means ± standard deviation. The effects between the groups and within the groups (baseline vs. 3d post-AMI) were analyzed by two-way analysis of variance (ANOVA) with repeated measures model with Bonferroni correction. The mean differences between the groups were detected by independent Student’s t-test. Differences were considered statistically significant at P<0.05. Statistical analyses were performed with SPSS software (version 17.0; Macintosh; SPSS IBM).
Reperfused AMI did not enhance the release of SDF-1α early (at 3 days) post-AMI (baseline: 32.02±24.35 vs. 3 days post-AMI: 26.97±15.43pg/ml; P=0.41) (Figure 3). In contrast, the circulating level of CXCR4 (baseline: 0.47±0.22 vs. 3 days post-AMI: 1.15±0.95ng/ml; P=0.034) significantly increased at 3 days post-AMI with concomitant induced mobilization of CD34+ cells (baseline: 260±75 vs. 3 days post-AMI: 668±180cells/μl; P<0.001). However, the level of MMP-2 was increased significantly (baseline: 291.83±53.40 vs. 3 days post-AMI: 369.64±72.88pg/ml; P=0.011), which might explain the cleaved SDF-1α/CXCR4 axis (Figure 3).
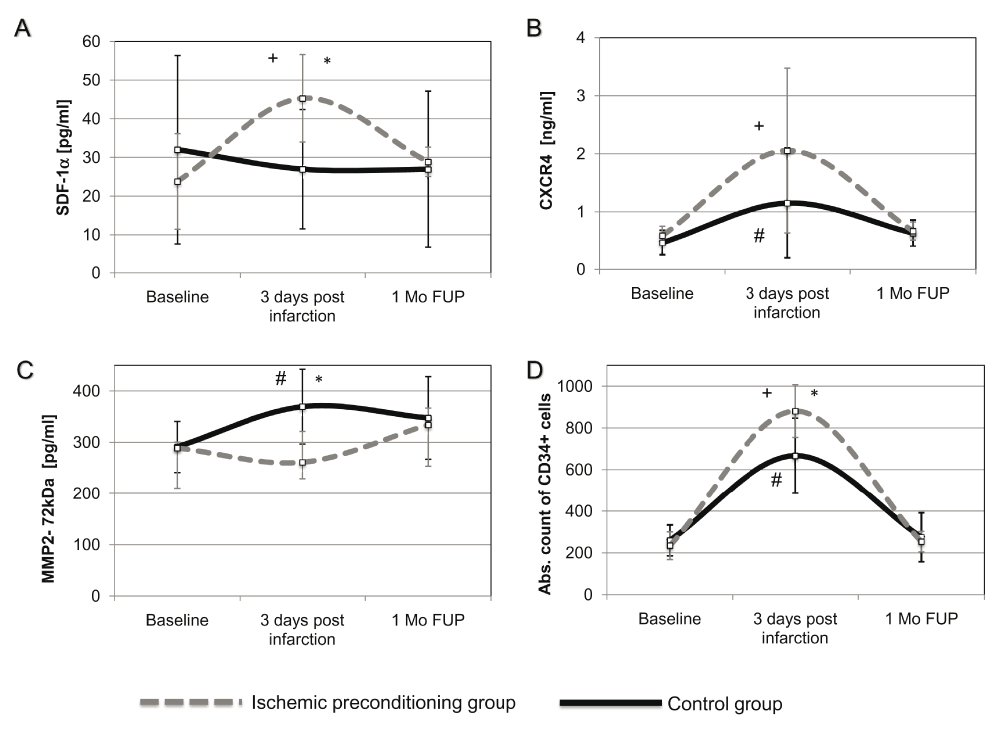
Plasma concentrations of circulating (A) SDF-1α; (B) CXCR4; (C) MMP-2 (72kDa) protein isoform measured by porcine-specific ELISAs. (D) Absolute count of circulating CD34+ cells were determined by FACS analysis. Concentrations are expressed as mean± standard deviation. *P<0.05 between the IP and control group; +P<0.05 between baseline and 3 day values within the IP group; #P<0.05 between baseline and 3 day values within the control (non-conditioned AMI) group. SDF-1α, stromal-derived factor-1 alpha; CXCR4, C-X-C motif chemokine receptor 4; MMP-2 (72kDa), matrix metalloproteinase-2, 72kDa isoform; IPC, ischemic preconditioning; AMI, acute myocardial infarction; 1 Mo FUP, 1 month follow-up.
The circulating levels of the angiogenic cytokines (FGF-2, VEGF, IL-8 and TNFα) were not changed significantly at 3-days post-AMI in the AMI group (Figure 4A–D).
IP led to the significantly higher stimulation of SDF-1α chemokine release with its putative receptor, CXCR4, into circulation, accompanied by downregulation of MMP-2. The number of CD34+ cells significantly increased as compared to the animals in the control group (non-conditioned AMI).
The plasma level of SDF-1α significantly increased 3 days post infarction in the IP group as compared to control AMI group (IP: 45.29± 11.31 vs. control: 27.00±15.43pg/ml; P=0.037), with normalization at the 1-month FUP (IP: 28.87± 3.81 vs. control: 26.91± 20.24pg/ml; P=0.85) (Figure 3A). Enhanced SDF-1α secretion was accompanied by significant increase of its soluble CXCR4 receptor after 3 days post-AMI (baseline: 0.59±0.16 vs. 3 days post-AMI: 2.06±1.42ng/ml; P=0.034); however, this did not reach statistical significance between the groups (IP: 2.06 ±1.42 vs. control: 1.15 ±0.95ng/ml; P=0.79) (Figure 3B).
IP significantly downregulated the secretion of MMP-2 into plasma at 3 days FUP as compared to the control AMI group (IP: 165.67±47.99 vs. control: 369.64±72.89pg/ml; P=0.004), which returned to the baseline level at the 1-month FUP control (IP: 334.00±93.10 vs. control: 347.58±80.47pg/ml; P=0.074) (Figure 3C).
FACS analysis was performed to reflect the impact of chemoattractant release on cell migration. We observed a significant parallel increase of mobilized CD34+ cells in both non-conditioned AMI and IP groups 3 days post infarction (IP: 881±126 vs. control: 668±180cells/μl; P=0.026, returning to the baseline level after 1 month FUP (IP: 255±50 vs. control: 275±118cells/μl; P=0.85) (Figure 3D).
A trend towards increase of IL-8 was observed in the IP group at 3 days and 1 month post infarction (IP: 100.18±60.42 vs. control: 49.52±16.68pg/ml; P=0.055) at day 3 and (IP: 59.32±32.88 vs. control: 25.19±5.76pg/ml; P=0.059) at 1 month (Figure 4A).
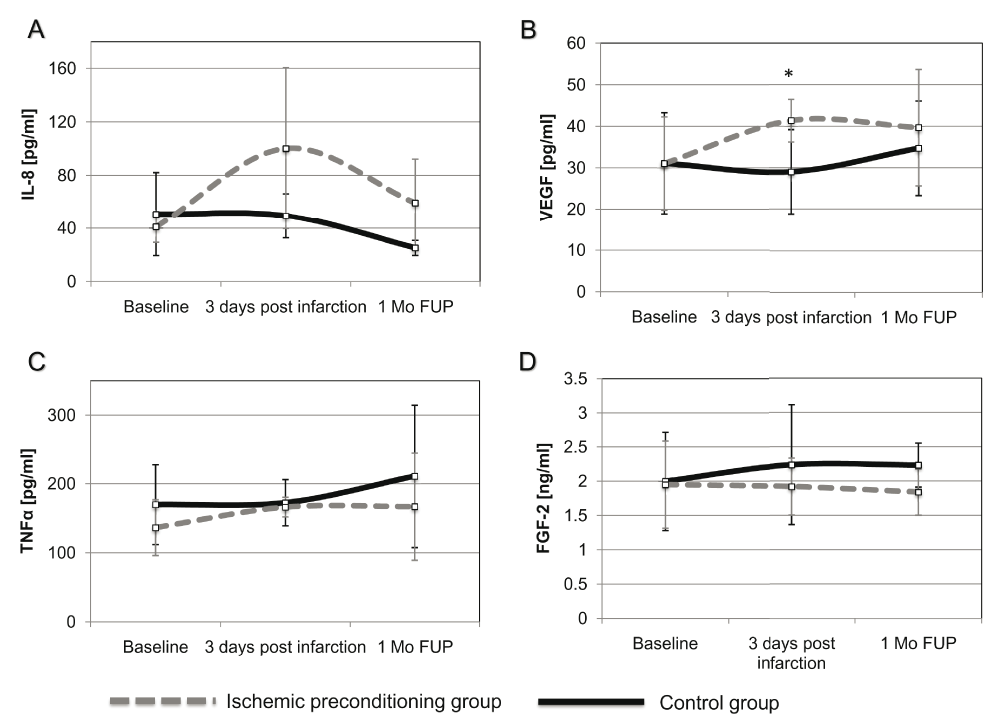
Plasma concentrations of circulating (A) IL-8, (B) VEGF, (C) TNFα, (D) FGF-2 Concentrations are expressed as mean± standard deviation. *P<0.05 between the IP and control group. FGF-2, fibroblast growth factor-2; VEGF, vascular endothelial growth factor; IL-8, interleukin-8; TNFα, tumor necrosis factor alpha; IPC, ischemic preconditioning; AMI, acute myocardial infarction; 1 Mo FUP, 1 month follow-up.
The concentration of VEGF in plasma significantly increased in IP group at day 3 post infarction as compared with controls (IP: 41.35±5.12 vs. control: 29.01± 10.18pg/ml; P=0.021) (Figure 4B).
IP did not affect the changes in serum concentrations of TNFα as compared to the control group (Figure 4C).
The plasma level of FGF-2 was not significantly changed at day 3 by IP as compared to the control group (IP: 1.90±0.41 vs. control: 2.22±0.88ng/ml; P=0.45) (Figure 4D).
In order to prove the oppositional effect of MMP-2 on CD34+ cell mobilisation, we added MMP-2 to cultured HACMs, stimulated with SDF-1α, and quantified the CD34+ cell migration towards the HACMs (Figure 5).
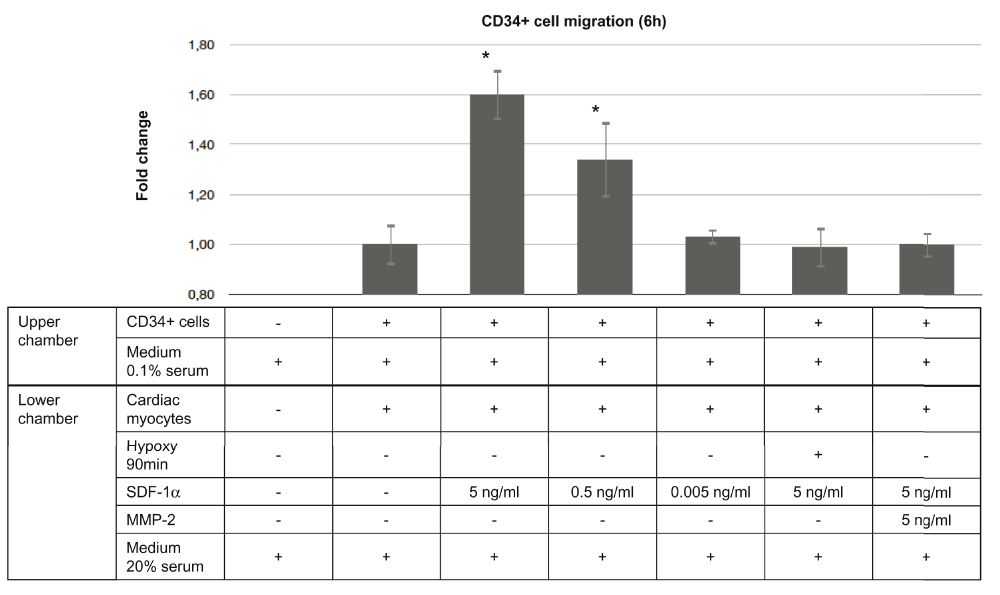
Migration was quantified as fold change impedance compared to the baseline conditions. Adding of SDF-1α in different concentrations induced chemotaxis of the CD34+ cells in a dose-dependent manner. A total of 90min hypoxia followed by a change of the medium eliminated the chemotactic effect of SDF-1α, and blocked CD34+ cell migration. This migration effect of SDF-1α was similarly eliminated if MMP-2 was added to the normoxic cell culture. Depicted results express impedance values measured at 6h post-treatment. Background results were subtracted from each impedance measurement. Parameters are expressed as mean ± standard deviation. Each experiment was repeated three times. *P<0.001 compared to the baseline normalized value. SDF-1α, stromal derived factor-1α; MMP-2, matrix metalloproteinase-2.
SDF-1α treatment stimulated the migration of CD34+ cells toward HACMs under normoxic conditions in a dose-dependent manner. The maximal chemotactic effect (1.6± 0.11 fold change; P<0.001) was achieved by adding 5ng/ml concentration of SDF-1α, while 0.5ng/mL and 0.05ng/mL SDF-1α resulted in a migration rate of 1.35± 0.18 fold (P<0.001) and 1.08±0.02 fold change (P=0.43), respectively, compared to the control HACMs and CD34+ cells culture without SDF-1α.
Co-incubation of HACMs with MMP-2 under normoxic conditions completely eliminated the SDF-1α chemotactic effect to CD34+ cell migration towards the cardiomyocytes (0.98±0.1 fold change; P=0.71).
Interestingly, incubation of the cardiomyocytes under 90min hypoxic conditions inhibited migration of CD34+ cells, even if the highest effective dose of SDF-1α (5ng/ml) was added to the cell culture as a chemoattractant (1.00±0.04 fold change; P=0.75).
Here, we demonstrate that 1) myocardial ischemia triggers the release of circulating MMP-2, which inhibits SDF-1α and CXCR4 release; 2) SDF-1-induced migration of CD34+ cells towards cardiomyocytes was inhibited by MMP-2 in vitro; 3) IP inhibited MMP-2 release, thereby increasing both SDF-1α and CXCR4 levels, resulting in a higher level of CD34+ cell mobilization 3 days post ischemic injury in in vivo condition; 4) IP induced VEGF secretion in the second window of cardioprotection.
Reperfused AMI led to an increase in CXCR4, but not SDF-1α, at 3 days post-infarction, with moderate enhancement of circulating CD34+ counts. Similarly, AMI caused significant elevation of MMP-2, produced by macrophages in case of acute tissue injury. MMP-2 disrupts the SDF-1α/CXCR4 axis, by cleaving SDF-1α to N-terminally truncated SDF-114. This form of SDF-1 is unable to trigger CXCR4 signalling and prevent the chemoattractant function of SDF-1α/CXCR4 in human progenitor cells15. Since the increased upregulation of MMP-2 post-AMI may inhibit retention of hematopoietic stem cells in the ischemic injury site, targeted modulation of MMP-2 expression has potential to improve outcome of regenerative therapies9.
Our previous study demonstrated that IP in early phase post-infarction (early window of protection, 2h after reperfusion start) induced mobilization of BM-derived haematopoietic (HSCs) and mesenchymal stem cells (MSCs) involving the release of distinct cytokines7. In our present work, we analyzed the effect of IP on the mobilization of CD34+ regenerative cells and measured the cytokine release (MMP-2, VEGF, FGF-2, IL-8 and TNFα) in the late (second) window of protection.
In contrast with the non-conditioned AMI group, we observed significantly elevated SDF-1α plasma level in the IP group at 3 days post infarction, as compared to the AMI group. This confirmed our earlier assumptions that SDF-1α is released in a later time window after IP7. Previous in vitro and in vivo experiments have shown an increased cell migration ability responding to treatment with SDF-1α16 or increased mobilisation of BM-derived cells toward injured tissue after SDF-1α overexpression6,17. The putative receptor for SDF-1α chemokine is CXCR4, which is expressed also in mouse cardiomyocytes17 and mobilises mesenchymal stem cells in the ST-segment elevation of myocardial infarction patients2. The elevated level of SDF-1α was paralleled by an increased number of circulating CD34+ cells. This suggests that IP stimulates CD34+ cell migration by SDF-1α/CXCR4 upregulation within the first days after AMI.
The increased concentration of MMP-2 (72kDa) at 3 days post-infarction was completely abolished by IP, which might be an additional beneficial effect of IP in a translational large animal model, and is similar to mice experiments18. IP has shown cardioprotective effects against ischemia/reperfusion injury in accepted experimental models. Induction of IP in a mouse model led to improvement of cardiac function and increasing cell survival, accompanied by release of BM- derived cells10. Accordingly, our previous4 and present study suggest that IP stimulates endogenous mechanisms, promoting the recruitment of CD34+ cells in both early and late windows of cardioprotection.
In order to prove the direct confounding effects of MMP-2 on SDF-1α/CXCR4, we have performed in vitro experiments, and observed that MMP-2 completely inhibited SDF-1α -induced CD34+ cell mobilization.
Interestingly, our experiments also revealed that 90min hypoxia abolishes the SDF-1α chemotactic effect in vitro. By contrast, it has been reported that hypoxia inducible factor 2, which is released in hypoxia, binds to the promotor sequence of CXCR4, the SDF-1α putative receptor, and activates the migratory activity of the endothelial progenitor cells19. We cannot completely explain our findings, but we assume that the release of hypoxia-triggered factors, such as MMP-2, may locally inhibit the migratory capacity of the regenerative cells. This is also in concordance with the findings in humans; early administration of regenerative cells has debatable effects on myocardial regeneration20.
Non-conditioned AMI did not influence the release of circulating cytokine FGF-2, VEGF, IL-8 and TNFα. In the first 3 days post-AMI. Our findings are similar to the study of Husebye et al21 reporting no increase in TNF-alpha and IL-8 levels in patients with STEMI and randomized to placebo group. In contrast, IP induced a marked release of circulating VEGF and a trend towards increase in IL-8 3 days post-AMI, indicating the stimulation of additional pro-migratory cytokines by IP for enhanced cardioprotection. IL-8 is a pro-inflammatory C-X-C chemokine that is also involved in activation of pro-angiogenic processes and re-introduction of progenitor cells into the circulation. The study of Schomig et al. demonstrated significantly increased IL-8 level in AMI patients as compared to patients diagnosed with stable angina8. In our study, we observed a trend toward increased release of IL-8 in the clinically relevant porcine reperfused “STEMI” model. The levels of CXCR4 increased both in controls (with AMI) and IP groups, with a trend towards higher increase in the IP group 3-day post AMI. The differences between our and other studies might be explained by the pre- and peri-AMI medication of patients with standard care that may contribute to changes in plasma levels of cytokines in AMI patients8. Results of plasma cytokine levels would be more informative if it measured more often, in an extended time window. The area under the curve (AUC) calculation of the cytokine release data might have delivered additional results. However, for a simple blood sampling, the animals must have been fully anaesthesized, which procedure signifies an additional stress for the animals with recent AMI with predicted higher mortality.
In our previous experiments, IP induced the release of VEGF plasma levels immediately after myocardial infarction (first window of protection)7. In the present experiment, VEGF was still increased 3 days post AMI in the IP group (second window of protection) as compared to the control AMI group. Similarly to our study, Kamota et al. showed an amplified secretion of VEGF and SDF-1α up until 6 hours post infarction in a mouse model of IP10. Tang et al. also reported induced mobilisation of stem cells by VEGF/ SDF-1α trafficking in a rat model6.
FGF-2 is an important chemotactic factor and it is also a prominent cardioprotective and angiogenic agent22. Since FGF-2 was not significantly induced by IP in our experiment, we assume that this protein did not participate in mechanisms of IP-elicited late window of cardioprotection.
Acute phase of AMI after IP is characterized by an increased level of TNFα triggering a release of additional cytokines, such as IL-6, IL-8, and cell adhesion molecules. Our previous data demonstrated that IP resulted in elevated levels of TNFα in serum with concomitant IL-8 increase immediately after reperfusion induction7. A later time window after AMI revealed heterogeneous results. TNFα remained moderately increased 3 days post infarction with continuous moderate increase after 1 month FUP in both groups, most probably due to developing chronic phase of myocardial infarction. Interestingly, IP induced a trend towards enhanced IL-8 release, which is a potent progenitor cell mobilisation enhancer responding to ischemia, although it is also associated with pro-inflammatory processes8,9.
In conclusion, the present study revealed that AMI induces MMP-2 release, which hampered the ischemia-induced increase in SDF-1α and CXCR4 by cleaving the SDF-1α/CXCR4 axis. This led to diminished mobilization of the angiogenic CD34+ cells. IP induced CD34+ cell mobilization in the late phase (second window), thereby also increasing circulating SDF-1α and CXCR4, parallel with enhanced VEGF secretion. One mechanism of this beneficial effect of IP might be the inhibition of AMI-induced MMP2-release. In vitro migration assay confirmed the anti-migratory effect of MMP-2 and the direct negative association of MMP-2 and SDF-1α-induced cell migration. Accordingly, our experiment might explain the inhibited homing of mobilized or transplanted cells in the ischemic myocardium resulting in decreased efficacy of cell-based therapies early after AMI.
Even though we demonstrate IP-induced mobilisation of CD34+ cells in a large animal model of reperfused AMI, the clinical relevance of IP remains uncertain. We have concentrated on mechanisms involved in cell mobilisation in terms of chemokine and cytokine secretion.
An important limitation is the utilisation of human CD34+ FACS antibody due to lack of commercially available porcine products. However, the number of mobilized CD34+ cells correspond with the available mobilized cell numbers published several times2,10,23; bearing in mind, that the normal count of white blood cells of pigs is 12–20 thousand cells/μl blood.
We revealed one additional possible beneficial mechanism of IP, namely the inhibition of MMP-2 release with consequent higher mobilization of CD34+ cells, which was confirmed in our in vitro experiment. However, a direct association between IP - MMP-2 - CD34+ axis had to be confirmed in vivo, by blocking MMP2 in animals subjected to AMI and IP. We have not measured myocardial MMP-2 level, which analysis would require harvesting of the animals maximal 72h post IP-AMI (second window of protection), and our animals survived 1-month follow-up.
We have chosen female pigs for the experiments because of a clear gender differences observed in female and male rodents, rabbits, dogs and pigs24; the incidence of cardiogenic shock and life-threatening arrhythmias were more frequent in male than female pigs by using the closed-chest reperfused AMI model12.
We are aware, that serial blood sampling would have given more information, e.g. the evident changes in mobilisation of bone marrow-derived cells following myocardial infarction occur at day 3, 7 and 14 post-ischemia25. However, we have focused on the second window of protection, which ends at day 3.
Inclusion of a sham-operated control group would promote better comparison of the results. However, we decided to not supplement the study groups with control group of animals without infarction with expected constant baseline level of biomarkers. Indeed, we intended to apply the “reduction” principle of the 3R (reduction, refinement and replacement) concept regarding the in vivo experiments.
Isolation of human cardiomyocytes was performed from samples obtained from both female and male patients. Since the cells were utilized for in vitro experimental evaluation, the “gender” of the cells in the cell culture was not relevant for the in vivo experiments.
Dataset 1. Raw data for XCelligence measurements of cell migration assay (DOI: 10.5256/f1000research.9957.d14207926).
Dataset 2. Raw data obtained from ELISA and FACS analyses (DOI: 10.5256/f1000research.9957.d14208027).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)