Keywords
Individualized Medicine, Clinical Genomics, Diagnostic Odyssey, Oncology, Molecular Modeling, Molecular Dynamics, Variants of Unknown Significance
This article is included in the Rare diseases collection.
Individualized Medicine, Clinical Genomics, Diagnostic Odyssey, Oncology, Molecular Modeling, Molecular Dynamics, Variants of Unknown Significance
In under a decade, sequencing of a human genome moved from a three billion dollar, multi institutional effort, to a common research assay. Now, 16 years after completion of the draft sequence, genomic testing is an increasingly prevalent component of clinical testing. Oncology, hematology and the diagnosis of rare genetic disorders (diagnostic odyssey) have particularly benefited from the increased genetic testing resolution afforded by modern sequencing technologies. The commercialization of clinical assays to profile patients’ somatic or germline genomes is creating the potential for higher resolution diagnoses and individualized treatment options. Stories of the resounding success of such efforts have been widely and justifiably publicized, and have now captured imaginations well beyond the laboratory or clinic; even so far as the White House and US Congress. The President’s Precision Medicine Initiative1 has now been signed into existence and guarantees expanded exploration within this burgeoning area of health sciences research.
For the researchers and clinicians on the ground of this nascent field, enthusiasm is high but it is tempered with frustration due to the as-yet high rate of cases for which genomic testing remains insufficiently informative (around 75% for diagnostic odyssey)2,3. While the explosion of genomics research has increased our understanding of the human genome and the wide-ranging functions it encodes, most regions of the genome remain uncharacterized and poorly understood. The majority of genomic sequence variants detected in clinical genomic testing go unreported or are classified as variants of unknown significance (VUSs) due to the lack of understanding of their potential to affect normal physiological function.
A recurring scene for many of the clinicians, genetic counselors and researchers who are adopting and exploring this new arena is the convening of large, multidisciplinary teams of clinical and research staff to pore over lengthy lists of genomic sequence variants, exchange professional opinion and debate next steps. However, in the end, these involved efforts often fail to establish variants with highly confident causal or mechanistic relationships with the disease phenotype. In oncology, the knowledge gained for treatment selection is often compelling, but with little prior evidence, most are difficult to actualize. For example, identification of a novel missense mutation in the functional domain of a known, druggable oncogene might logically appear to be a therapeutic target, but no information may exist linking the mutation to drug efficacy. It is this disconnect between novel genotypes identified and their link to the patient’s phenotypes which thwarts our ability to further improve clinical decision making. Thus, there is a significant need to overcome this critical challenge to expand the success of genomic testing in individualized medicine.
Wet-lab assays designed to ascertain functional relevance of specific mutations are a highly desired solution, but they remain cumbersome, time-consuming and cost prohibitive in the great majority of cases. They are therefore misaligned with the high throughput nature of modern genomics. Meanwhile, computational methods of predicting the pathogenicity of genomic sequence alterations exist and are both amenable to high-throughput predictions and widely applied in the field4. These algorithms utilize varied information including evolutionary conservation, genomic position or basic protein-level structural information to assign probabilistic scores or categorical predictions of pathogenicity. Their accuracy varies widely, with alternative tools often producing conflicting predictions for the same variant. Furthermore, the predictions often lack contextualization in terms of biological effect. Algorithmic categorization of a variant as pathogenic offers little insight into the nature of its phenotypic effect and less indication still of whether it is relevant clinically or how to act upon it. Many researchers continue to work to improve these methods, but the burden of VUS interpretation persists and the need for a means to address this burden and facilitate clinical interpretation is widely felt within the field.
The aforementioned computational tools largely used leverage genome centric information. While there is much proven value to these data, the biological reality comprises many additional layers of complexity. Mutations affect atomic-level biophysical changes that may alter the structure and biochemical function of the genome's protein products. While we detect variants in DNA, we typically interpret their effect by inferring or confirming their deleterious effect on encoded proteins (Figure 1). Even the effects of regulatory variants are typically interpreted as altering the probability of a protein to be expressed or spliced into a given functional form. Thus ideally, any method used to assess pathogenicity of genomic variants should possess the ability to look beyond genomic annotation to a functional context, at an atomic resolution. This increased resolution is likely to yield information that can add context to mutations, better identify the mechanism of pathogenicity, and combine with existing knowledge to facilitate in clinical decision making.
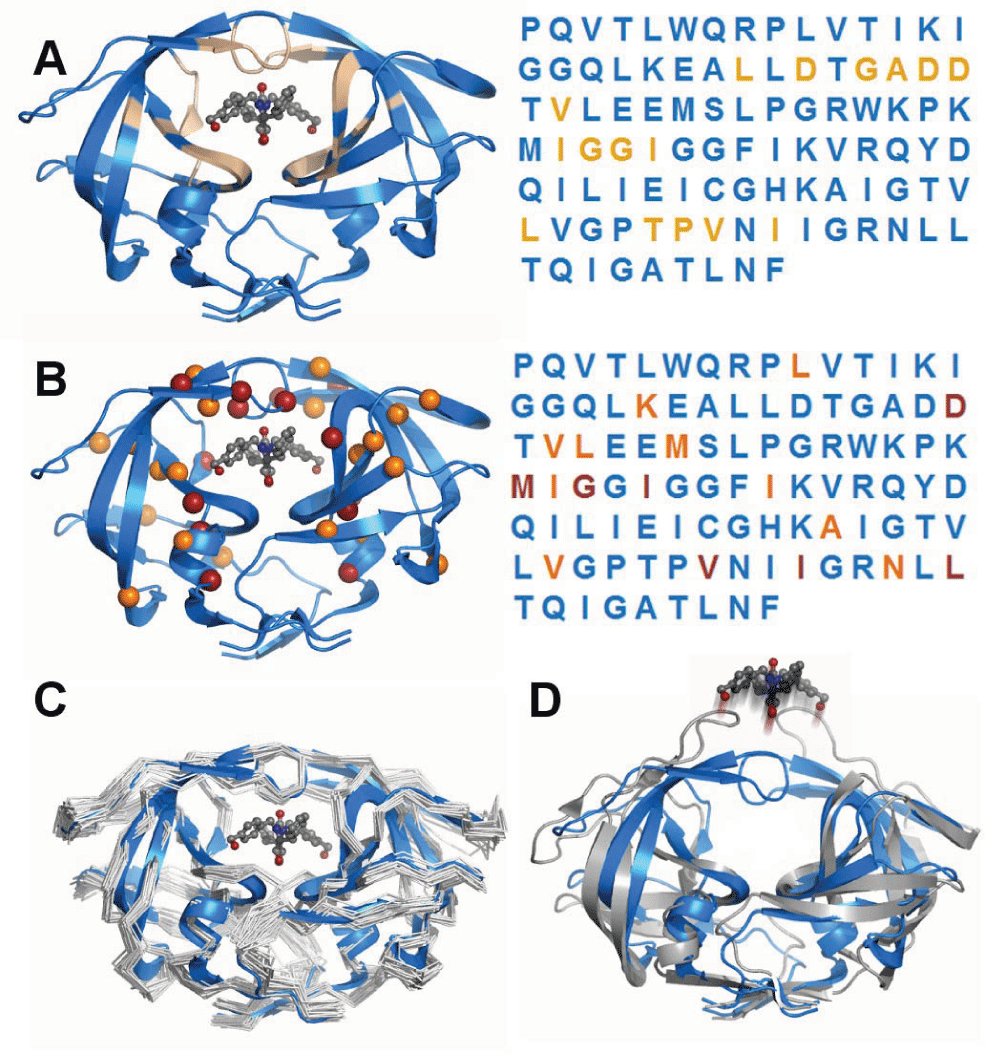
HIV-1 protease has become a model system because of its disease relevance, the availability of mutational and drug binding data, and for its tractable size and molecular stability. The protein’s function is to cleave HIV peptides into the functional proteins of the infectious HIV virion. A) Ligand binding residues are spatially separated. The functional protein dimer is shown with a pharmacologic inhibitor bound to the active site. Residues that are within 3.5Å of the inhibitor are highlighted in tan. The primary sequence is colored identically to the three-dimensional structure to indicate relative positioning of residues. It is apparent that the ligand binding portion of the protease consists of residues that are non-adjacent within the primary sequence, illustrating an advantage of modelling over linear sequence analysis. B) Drug resistance mutations tend to occur in residues within the active site. Many mutations have been characterized that are associated with resistance to inhibitor drugs. While the sites of these mutations are also disjoint in sequence, nearly all of them fall into the same set of drug binding residues, indicating how modeling can enable prediction of a mutation’s effect. C) Structural effects of mutations beyond the active site. Drug-resistance mutations in non-ligand-binding residues have been shown, using computational experiments, to impact the flexibility of the protein and therefore alter drug binding. Computational modeling has characterized the flexibility of the protein in multiple mutated states, illustrating the potential to predict the functional effect of mutations beyond an active site. D) Dynamics of unbound ligand. Computational studies have the advantage of being able to simulate conditions that are difficult to assay experimentally, such as the dynamics of ligand-free forms in atomic detail.
Motivated by the conditions described, we have begun to apply molecular modeling and dynamic simulation techniques in the interpretation of genomic variants identified by next generation sequencing. These methodologies abandon the practice of regarding mutations as occurring in linear strings of nucleotides or amino acids and instead offer a three dimensional, dynamic view, at an atomic resolution. They involve the computational generation, optimization and verification of a protein model, often based on homology to an experimentally determined protein structure5,6. Ab initio modeling is also possible, albeit with lower confidence. The model itself contains information about the linear amino acid sequence of the protein, along with the relative spatial coordinates of its atoms. Precise mathematical and biophysical parameters in the form of a force field are applied to the model to calculate energetic characteristics of the system. The methods are often mature and under reasonable conditions can be expected to produce a model with error comparable to that of a typical structure solved by nuclear magnetic resonance spectroscopy.
Molecular modeling allows us to visualize the manner in which proteins are folded to create a functional structure and to accurately simulate how this is disrupted by mutational events. Because we can visualize atomic bonds, mutations which disrupt inter-molecular interactions - for example between an enzyme and its substrate - can also be modeled. To illustrate their function, many enzymes are analogized to hand tools. One example likens proteases and scissors; if a variant inhibits the closing motion of the scissors about their fulcrum, the blades cannot function; if a variant blocks entry of a material between the blades, then that material can no longer be cut. The reality, of course, is more complex. Proteins are flexible polymers that only achieve mechanistic accuracy by folding into complex and specific three dimensional conformations that restrain or focus thermal fluctuations towards collective motions that are typically part of the mechanism itself. Regions of the properly folded structure’s surface that interact with other molecules, either for chemical modification (e.g. phosphorylation) or structural contacts (e.g. α/β tubulin assembly), can also be critical for function and modified by variants. In addition to the native shape of a protein, the ability of the linear amino acid polymer to achieve that shape is critical. Protein folding often occurs through progressive assembly of local structural elements or intermediates. If an intermediate is either stabilized or destabilized by a variant, the ability of the protein to achieve the native fold could be altered.
Computational biophysics and biochemistry aim to understand molecular function in a dynamic manner at an atomic resolution differing from their wet-lab counterparts methodologically but sharing the same goals. The most obvious advantages of computational approaches include the potential to test hypotheses in silico that would be difficult, costly, or intractable in the lab. Similar to laboratory experiments, computational calculations and simulations are most interpretable when they are well designed, test a specific hypothesis, contain positive and negative controls, connect the data generated to the pre-existent knowledge in the field, and allow drawing further functional inferences. When all these conditions are met, the three dimensional and dynamic representation of the modeled mutations may add a significant value to the interpretation of a genomic test’s finding.
Of course, molecular modeling methods have limitations. The generation of a reliable model is often dependent on the pre-existence of experimentally determined homologous structures and even where these exist, they may provide only partial information7. A model is by nature an approximation of reality. Nonetheless, current techniques have achieved suitable accuracies such that they are frequently used8–11 in applications including drug design, virtual screening, protein engineering and site-directed mutagenesis. With this in mind, their relatively slow uptake in the clinical setting is somewhat surprising. This fact may simply reflect the relative nascence of clinical genomics and the tendency of specialists to seek out fields in which their specialty is already known and accepted.
We encourage those working within or in proximity to the clinical genomics setting to engage in or promote increased exploration of these methods in their work. Our initial experiences of applying such techniques at the clinical-research boundary of genomics-driven oncology, hematology and diagnostic odyssey have been encouraging and have begun to inform decision-making. We are observing clear initial benefits in regard to variant prioritization, interpretation and validation, with several initial publications in preparation to highlight the value obtained. Of course, not everyone will possess the necessary scientific knowledge or technical skills to deploy these methods directly, but we propose that inter or intra-institutional collaborations may enable those lacking the requisite expertise to identify and access appropriate resources. Furthermore, several freely available online solutions exist12,13 and enable a researcher or clinician to experiment with core modeling methods in the absence of extensive technical expertise. Such independent or collaborative exploration will open the door to deeper understanding of biological mutations and have the potential to inform clinical thinking.
In summary, genomic testing is assuming an increasingly prominent role in the identification, prioritization, and interpretation of disease-associated genomic variants. While a few of these variants are known to be pathogenic, knowledge of the deleterious effects of the majority remain elusive. Laboratory methods remain gold-standards for functional characterization, but are generally incompatible with large-scale characterization of variant effects. Predictive algorithms are in some instances successfully applied to differentiate pathogenic variants from variants of unknown significance. However, these methods largely ignore measures of protein structure, energies, molecular bonds, intermolecular interactions, post-translational modification effects, protein aggregation, and stability. Conversely, this information lies at the heart of molecular modeling and dynamic simulation, which collectively equip us more fully to grapple with interpreting the effects of VUSs. We strongly believe that these methodologies constitute the future frontiers in forging the analytical pipeline of interpreting the results of genomic testing for individualized medicine and advocate their increased deployment within variant characterization efforts.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (1)