Keywords
Hi-C, ChIA-PET, infrastructure, data representation, genomic interactions
This article is included in the Bioconductor gateway.
This article is included in the RPackage gateway.
Hi-C, ChIA-PET, infrastructure, data representation, genomic interactions
This new version fixes some small typographical errors and clarifies the R version requirements for the S4 class framework.
See the authors' detailed response to the review by Nicolas Servant
See the authors' detailed response to the review by Douglas Phanstiel
Techniques such as chromatin conformation capture with high-throughput sequencing (Hi-C)1 and chromatin interaction analysis with paired-end tags (ChIA-PET)2 are increasingly being used to study the three-dimensional structure and organisation of the genome. Briefly, genomic DNA is fragmented and subjected to a ligation step during which DNA from interacting loci are ligated together. High-throughput paired-end sequencing of the ligation products will identify pairs of interacting genomic regions. The strength of each interaction can also be quantified from the number of read pairs connecting the two interacting regions. This information can be used to derive biological insights into the role of long-range interactions in transcriptional regulation as well as the general organization of the genome inside the nucleus.
The analysis of Hi-C and ChIA-PET data is not a trivial task, and many software packages have been developed to facilitate this process. Several of these packages like diffHic3 and GenomicInteractions4 are part of the open-source Bioconductor project, which aims to provide accessible tools for analyzing high-throughput genomic data with the R programming language. One of the strengths of the Bioconductor project is the quality and quantity of shared infrastructure available to developers5. Pre-defined S4 classes such as GenomicRanges and SummarizedExperiment can be used to represent various types of genomic data and information, easing the maintenance burden for developers while also improving interoperability between packages for users. However, this kind of common infrastructure does not yet exist for the genomic interaction field. Instead, each package contains its own custom classes, which increases code redundancy and development load while reducing interoperability.
Here, we describe the InteractionSet package that provides base S4 classes for representing and manipulating genomic interaction data. It contains the GInteractions class, to represent pairwise interactions; the InteractionSet class, to store the associated experimental data; and the ContactMatrix class, to represent interactions in a matrix format. This facilitates code reuse across Bioconductor packages involved in analyzing data from Hi-C, ChIA-PET and similar experiments.
Each object of the GInteractions class is designed to represent interactions between pairs of “anchor” regions in the genome (Figure 1A). It does so by storing pairs of anchor indices that point towards a reference set of genomic coordinates (specified as a GenomicRanges object). Each anchor index refers to a specific reference region, such that a pair of such indices represents a pairwise interaction between the corresponding regions. This design reduces memory usage as the reference coordinates need only be stored once, even if each region is involved in multiple interactions. Computational work is also reduced as calculations can be quickly applied across the small set of reference regions, and the results can be retrieved for each interaction based on the anchor indices. In addition, the GInteractions class inherits from the Vector class in Bioconductor’s S4Vectors package. This allows storage of metadata for each interaction (e.g., intensities, p-values) and for the entire object (e.g., experiment description).
The InteractionSet class is designed to store experimental data for each feature (Figure 1B). It inherits from the SummarizedExperiment base class, where each object of the class stores any number of matrices of the same dimensions. Each row of each matrix corresponds to a pairwise genomic interaction (represented by a GInteractions object that is also stored within each InteractionSet object), while each column corresponds to an experimental sample. Each entry of the matrix then represents the observation for the corresponding interaction in the corresponding sample. Different matrices can be used to store different types of data, e.g., read counts, normalized intensities. The InteractionSet class also inherits a number of fields to store metadata for each interaction, for each sample, and for the entire object.
The ContactMatrix class is designed to represent pairwise interactions in a matrix format (Figure 1C). Each row and column of the matrix represents a genomic region, such that each cell of the matrix represents an interaction between the corresponding row/column regions. Experimental data for that interaction can be stored in the associated cell. This provides a direct representation of the “interaction space”, i.e., the two-dimensional space in which (x, y) represents an interaction between x and y. Like the GInteractions class, the genomic coordinates are not stored directly – rather, the rows/columns have indices that point towards a reference set of coordinates, which reduces memory usage and computational work. The matrix representation itself uses classes in the Matrix package to provide support for both dense and sparse matrices. The latter may be more memory-efficient, particularly for sparse areas of the interaction space. ContactMatrix instances can also be easily converted to instances of existing matrix-based classes such as those in the HiTC package6.
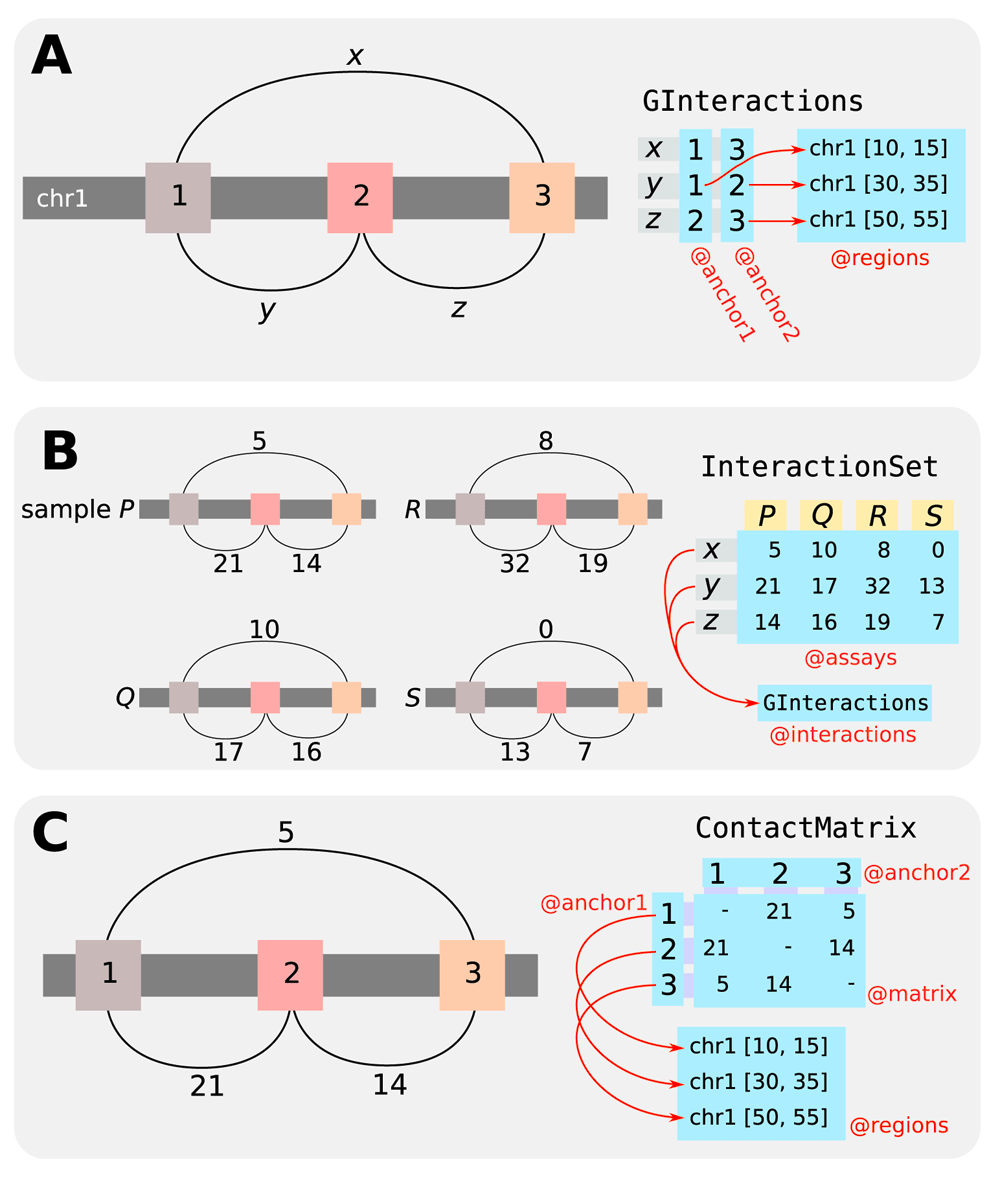
Relevant slots of each class (i.e., data values stored in each object of the class) are labelled with a preceding “@”. (A) The GInteractions class represents pairwise interactions between genomic regions by storing pairs of anchor indices that refer to coordinates in a GenomicRanges object. (B) The InteractionSet class stores experimental data in an “assays” matrix where each row is an interaction and each column is a sample. Here, counts represent the number of read pairs mapped between each pair of interacting regions in each sample. (C) The ContactMatrix class represents the interaction space as a matrix, where each cell represents an interaction between the corresponding row/column regions.
The InteractionSet package provides a variety of methods for manipulating objects of each class. In addition to slot accessors and modifiers, methods are available to convert objects to different classes in the same package (e.g., GInteractions to ContactMatrix) or to base Bioconductor classes (e.g., GInteractions to GRangesList). The distance between anchor regions on the linear genome can be computed for each pairwise interaction, to use in fitting a distance-dependent trend1 for diagnostics or normalization. The minimum bounding box in the interaction space can also be defined for a group of interactions (Figure 2A) to summarize the location of that group.
The InteractionSet package supports one- or two-dimensional overlaps for its objects (Figure 2B). A one-dimensional overlap is considered to be present between an interaction and a genomic interval if either anchor region of the interaction overlaps the interval. This can be used to identify interactions overlapping pre-defined regions of interest. A two-dimensional overlap is considered to be present between an interaction and two genomic intervals if one anchor region overlaps one interval and the other anchor region overlaps the other interval. This can be used to identify interactions linking two specific regions of interest, e.g., a gene and its enhancer. The same framework can be used to define two-dimensional overlaps between two interactions, based on whether the corresponding anchor regions overlap – this can be used to relate similar interactions in different GInteractions objects or across different experiments. More generally, interactions can be identified that link any two regions in a set of regions of interest. For example, given a set of genes, interactions between two genes can be identified; or given a set of genes and another set of enhancers, interactions linking any gene to any enhancer can be found.
Hi-C data in an InteractionSet object can also be converted into a 4C-like format (Figure 2C). Firstly, a bait region is defined as some region of interest, e.g., a target gene or enhancer. All interactions in the InteractionSet object that have one-dimensional overlaps with the bait are identified. For each overlapping interaction, the anchor region that does not overlap with the bait is extracted and – along with the data associated with that interaction – used to construct a RangedSummarizedExperiment object. This process yields data for intervals on the linear genome, which is similar to the output of 4C experiments7 that measure the intensity of interactions between the bait and all other regions. The “linearized” format may be preferable when a specific region can be defined as the bait, as intervals on the linear genome are easier to interpret than interactions in two-dimensional space.
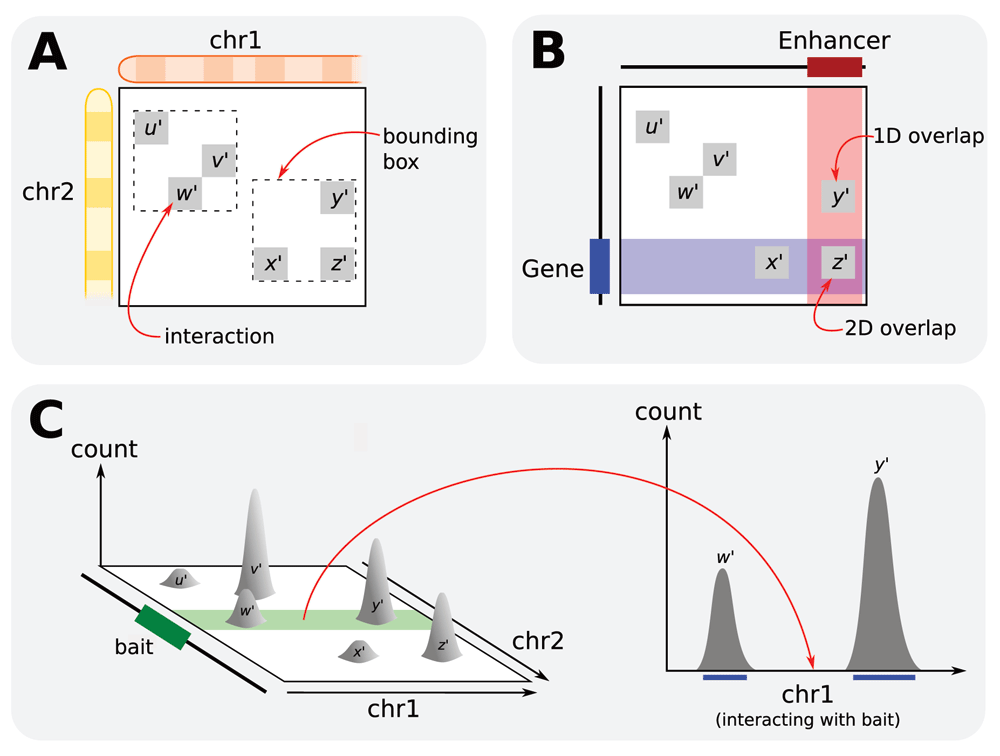
(A) Minimum bounding boxes can be identified for groups of interactions using the boundingBox method. Here, u′, v′ and w′ belong in one group while x′, y′ and z′ belong in another. (B) One- or two-dimensional overlaps can be identified between interactions and one or two genomic intervals, respectively, using the findOverlaps method. Here, x′ and y′ have one-dimensional overlaps with the gene and enhancer, respectively, while z′ has a two-dimensional overlap with the gene and the enhancer. (C) An InteractionSet object contains data – in this case, read pair count data – for interactions in the two-dimensional interaction space. Given a bait region, a “cross-section” of the space can be extracted and converted into a RangedSummarizedExperiment object using the linearize method. This object holds count data for intervals on the linear genome (blue lines) where the count for each interval describes the strength of the interaction between that interval and the bait. This format effectively mimics that of 4C data.
All classes and methods in the InteractionSet package are implemented using the S4 object-orientated framework in R (version 3.3.0 or higher). Classes are exported to allow package developers to derive custom classes for their specific needs. Pre-existing Bioconductor classes and generics are used to provide a consistent interface for users. After loading the InteractionSet package into an R session, instances of each class can be constructed from existing data structures, either directly (e.g., GInteractions objects from GRanges via the GInteractions constructor, or from Pairs via the makeGInteractionsfromGRanges function; ContactMatrix objects from GRanges and Matrix via the ContactMatrix constructor) or in a hierarchical manner (e.g., InteractionSet objects from matrices and a GInteractions object via the InteractionSet constructor). The methods described above can then be applied to each instance of the class. While the InteractionSet package does not have functions to load data from file, it can be combined with the import function in the rtracklayer package8 to construct class instances after importing data from a range of formats including BED and BEDPE. A similar strategy can be used to export data to file.
The availability of common infrastructure is highly beneficial to software development by reducing redundancy and improving reliability, as more developers can check the same code; improving interoperability, as different packages use the same classes; and increasing the accessibility of useful features, which exist in a single package rather than being sequestered away in a variety of different packages. Here, we present the InteractionSet package that implements a number of classes and methods for representing, storing and manipulating genomic interaction data from Hi-C, ChIA-PET and related experiments. The package is fully integrated into the Bioconductor ecosystem, depending on a number of base packages to implement its classes (e.g., S4Vectors, GenomicRanges, SummarizedExperiment) while in turn being depended on by packages for higher-level analyses (e.g., diffHic, GenomicInteractions). Indeed, for any new packages, use of the features in InteractionSet will simplify development and improve interoperability with existing packages in the Bioconductor project. The InteractionSet package itself can be obtained for R version 3.3.0 at http://bioconductor.org/packages/InteractionSet.
Software and latest source code available from: http://bioconductor.org/packages/InteractionSet
Archived source code as at time of publication: http://dx.doi.org/10.5281/zenodo.512049
License: GNU General Public License version 3.0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)