Keywords
SuperPC, binary outcome, continuous, time-to-event, forest plot,
This article is included in the RPackage gateway.
SuperPC, binary outcome, continuous, time-to-event, forest plot,
Heterogeneity in terms of tumor characteristics, prognosis, and survival among cancer patients has been a persistent problem for many decades. Currently, prognosis and outcome predictions are solely based on clinical factors. However, inaccurate prognosis and prediction may result from using only these. For example, based on clinical factors only, two tumor patients may have the same prognosis, but may not respond to the same treatment as the tumors may have a completely different molecular composition1–5.
One way to improve prognosis and clinical outcome predictions in cancer is to examine such associations using a tumor’s genomic profile. To achieve this goal, Bair and Tibshirani6 proposed a supervised principal component (SuperPC) method to identify subtypes of cancer. The method uses both gene expression and clinical data to diagnose future patients. This approach was applied to several publicly available datasets, showing that it was able to accurately predict the clinical outcome of interest based on a given gene expression profile. Since its inception, SuperPC has been introduced as a powerful tool for cancer diagnosis and treatment.
Currently, the SuperPC method has been developed as an R package, ‘superpc’ but as it stands, it is unable to address the following:
1) clinical association based on a binary outcome (e.g. responders versus non-responders);
2) ease of use for clinicians and researchers with limited programming skills; and
3) a visual summary of results.
To address these limitations, we developed GAC: Gene Association with Clinical, an interactive, GUI-based web-based application for analysis of gene associations with various clinical outcomes of interest. We developed GAC based on the R packages ‘shiny’ and ‘superpc’. Our GAC tool enables the user to perform a SuperPC analysis for three types of outcomes: time-to-event, continuous, and binary, and provides a summary of results using forest plots that may be readily exported into a file.
SuperPC is a generalization of principal component analysis, which generates a linear combination of the features or variables of interest that capture the directions of largest variation in a dataset. Instead of using the whole dataset directly, SuperPC defines a list of genes based on their association with an outcome of interest. To select the list of genes, a univariate score for each gene is calculated and those features (a.k.a., genes) whose score exceeds a threshold are retained as input into a principal component analysis, based on the retained features. For details, refer to Bair and Tibshirani6.
SuperPC for time-to-event was conducted using the ‘superpc’ package in R. Depending on the sample size of the original dataset; the researcher selects what proportion of the dataset to split into training and testing. The researcher can also specify how many numbers to test to check which the optimal threshold is. The number of folds for cross validation to determine the threshold also needs to be determined. There is also an option to run the analysis randomly, or upload fold IDs to replicate an analysis that was previously carried out. The association between the time-to-event outcome and the predicted principal component may be represented in a KM plot by dichotomizing the principal component using the median (Figure 1).
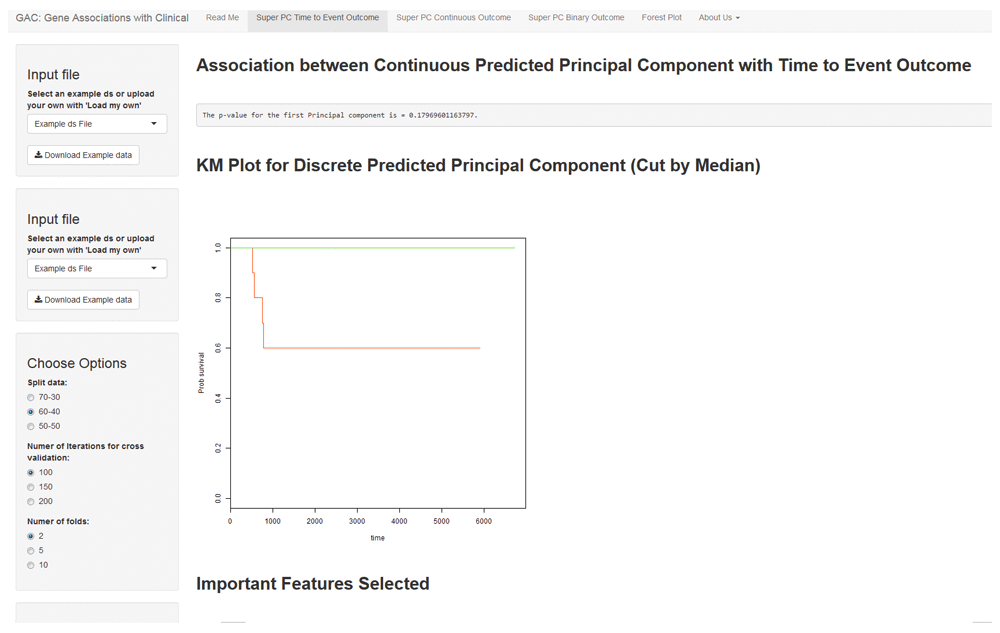
The interface shows an example SuperPC for time-to-event outcome. The left panel allows the user to select the various options such as data split, the optimal threshold, number of folds and a choice of generating new fold IDs or using a pre-existing set to replicate results. The right panel includes the results of the analysis. The KM plot displays the association of the outcome with the predicted principal component by the median. In addition, the univariate analysis regression scores and tables are also available for download.
SuperPC for continuous outcomes is implemented using the ‘superpc’ package in R, with the same options as time-to-event analysis. The predicted principal component is presented visually as continuous values through a scatter plot along with Pearson’s correlation (Figure 2). The predicted principal component could also be presented as binary groups (cutoff at median) through a boxplot, with a t-test applied.
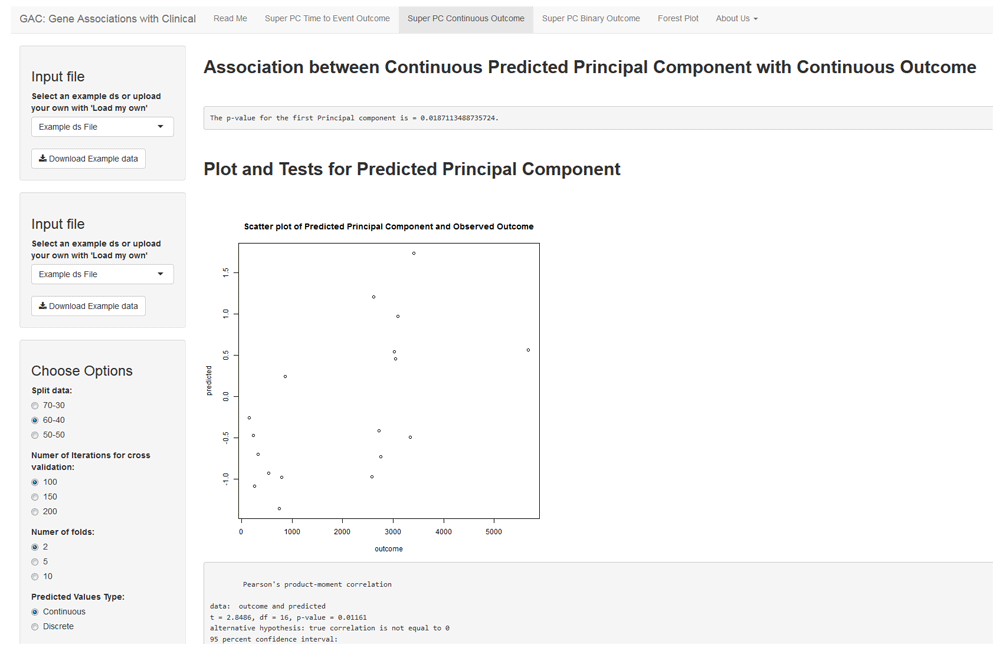
The interface shows an example SuperPC for continuous outcome. Similar to Figure 1, the user can choose the appropriate settings using the left panel and view the results of the analysis to the right. The association of the continuous outcome with the predicted principal component is summarized using a scatter plot (as seen). The user can alternatively choose to summarize these results through boxplots by dichotomizing the predicted principal component based on the median. As in Figure 1, the univariate regression scores and plots are available for download in the left panel.
SuperPC for binary outcome follows the same analysis workflow as the other two outcomes. The predicted principal component can be visualized as either a continuous variable through a box plot, with a t-test to summarize the statistical association (Figure 3), or as binary groups (cutoff at median) using a bar plot, with a chi-square test to summarize the statistical association between the predicted and the observed outcome.
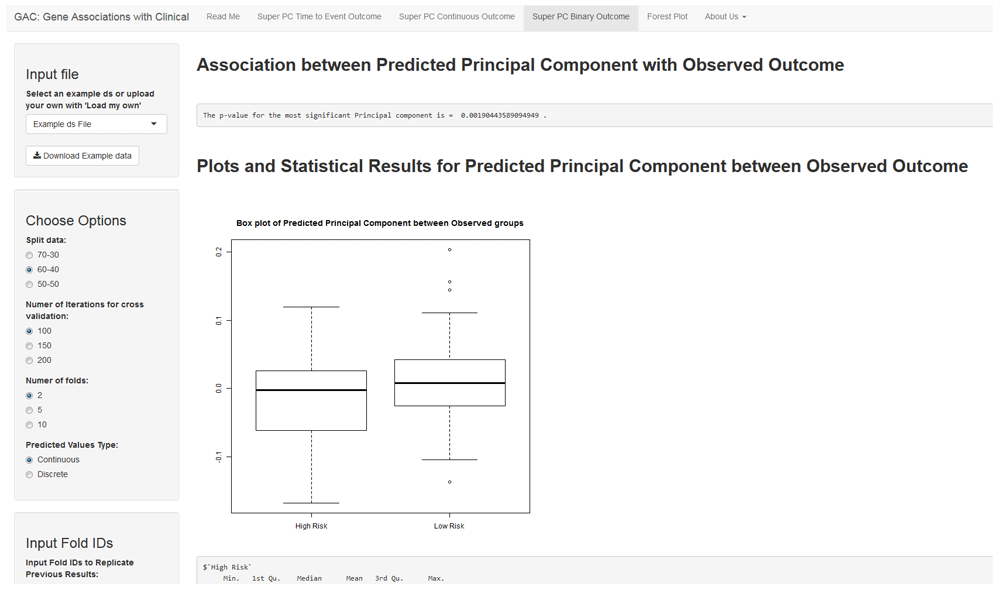
The interface shows an example SuperPC for binary outcome. The user can opt for similar options as in the previous figures. Also, as in Figure 2, the user has a choice to display the continuous predicted principal component as a scatter plot, or divide it into binary discrete groups (using median cut-off) to represent the association through a barplot. Similar download options are available.
A forest plot is a graphical display of point estimates of association widely used in meta-analysis. It has become popular for displaying the associations between clinical and genomic data. With our GAC tool, users have the option to generate a forest plot to display results (Figure 4).
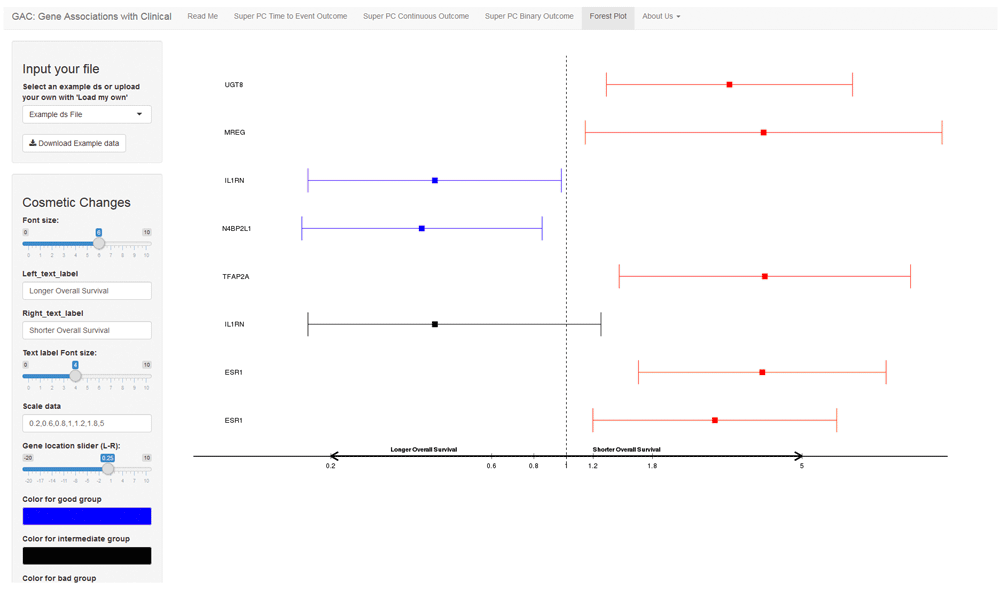
The interface shows an example forest plot. The left side comprises a user menu and the right includes the result plot. Users can upload their own summarized results with hazard ratio and confidence intervals for the survival outcome, or odds ratio and confidence intervals for the binary outcome. For graphical display, the researchers could choose to input different labels, font sizes and colors.
The GAC tool is written in R and tested using version 3.3.0. The interactive plots and data tables are made available using the shiny R package (www.rstudio.com/shiny).
Using a windows 7 Enterprise SP1 PC with a 32.0 GB RAM and a 3.30 GHz Intel® Xeon® Processor E5 Family, the time-to-event and regression analysis with 45 patients from the TCGA BRCA dataset was completed in 2.54 s and 1.80 s, respectively. The binary outcome analysis using the CoMMPass data of 135 patients was completed in 65.05 s. Source code is available at https://doi.org/10.5281/zenodo.803309.
GAC is a suite of tools that allows the user to conduct statistical analysis to identify and visualize the association between clinical outcomes of interest and genomic data using an interactive application in R.
For SuperPC time-to-event and regression analysis, we used TCGA BRCA RNASeqV2 gene expression and clinical data, downloaded from the TCGA data portal (now accessed at https://portal.gdc.cancer.gov/)7. The data included 380 differentially expressed genes when favorable (patients who did not die with at least 7 years of follow up) and unfavorable (patients who died 30 months post-treatment) outcomes from 45 patients were compared.
For SuperPC binary outcome analysis, CoMMPass IA9 RNASeq expression and clinical data was downloaded from the publicly available Multiple Myeloma Research Foundation (MMRF) database (https://research.themmrf.org/rp/download). Patient cytogenetics for outcome dichotomization was obtained from the MMRF data portal’s ‘Analysis Tools’ section (https://research.themmrf.org/rp/explore/). 135 patients with clinical, gene expression and copy number data were classified as high risk based on cytogenetics and the remaining 343 as not high risk. Among these patients, the top 1450 most variable genes were used.
The example data used in this article is available in Zenodo (https://doi.org/10.5281/zenodo.803309).
License: GAC is available under the GNU public license (GPL-3).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)