Keywords
nanopore sequencing, MinION, \textit{de novo} sequencing, review
This article is included in the Nanopore Analysis gateway.
nanopore sequencing, MinION, \textit{de novo} sequencing, review
The development of novel genome sequencing methods has been a major driving force behind the rapid advancements in genomics of the last decades. Notably, the advent of second generation sequencing (SGS) provided researchers with the required throughput and cost-efficiency to sequence many more genomes than was previously deemed feasible. Recent years saw the dawn of what can be perceived as a third generation; one that allows amplification-free reading of single DNA molecules in long consecutive stretches [van Dijk et al., 20141, review.] Currently, this new generation is dominated by two methods: nanopore sequencing and single-molecule real time (SMRT) sequencing, championed by Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio), respectively.
Conceptually, nanopore sequencing is easier to explain than most other sequencing methods. An electrical potential is applied across an insulating membrane in which a single small pore is inserted. A DNA strand is pulled through the pore and the sequence is inferred from the characteristic way in which the passing base combinations influence the current. In 1989, David Deamer roughly sketched this concept as it is applied today, although it took more than two decades of key innovations to bring the concept to fruition2. Since the introduction of the first commercially available nanopore sequencing device, ONT’s MinION, and the start of the MinION Access program (MAP) in 2014, the field of nanopore sequencing has been advancing at a rapid pace; both new applications and improvements to existing ones are published on a regular basis.
The advantages of the MinION over other sequencing devices are numerous. Both its size, roughly that of a cellphone, and its cost, a thousand dollars for a starter kit, are a mere fraction of that of competitors. Running the MinION is also reasonably time- and cost-effective; an 48-hour sequencing run currently costs about 800 dollarsi and typically yields about 5 Gbase3, although recently yields as high as 15.7 Gbase have also been reported (Baptiste Mayjonade, personal communication July 6th, 2017). Furthermore, the technique does not rely on any labeling techniques to recognize different bases, while Sanger, SGS methods and SMRT do require some form of labeling of nucleotides. Amplification by PCR is optional for the MinION, while this step is mandatory for Sanger and SGS-methods. Not only does omitting these steps simplify sample preparation for MinION samples, it also helps to avoid errors and biases (e.g. the CG-bias for PCR) and allows detection of modified bases4. Finally, the maximum read length produced by the MinION is many times greater than that of both second-generation and Sanger sequencing and only paralleled by SMRT sequencing, which is highly advantageous in resolving repeat sequences.
The most prominent disadvantages of the MinION, with respect to its competitors, are the lower signal-to-noise ratio, stochasticity introduced by its biological components, and the resulting high error rate of basecalling. Indeed, the MinION is a product in development and the used materials (i.e. membranes, nanopores and buffers) are still being optimized. Furthermore, it is thought that significant improvements are still possible in the software pipelines that translate current signal to DNA sequence, even though recent efforts have increased the base-calling accuracy dramatically, from around 85% in the first half of 20165–7 to over 95% in March of 2017.
In this review, an up-to-date overview of several aspects of nanopore sequencing is provided. First, the physical sequencing process as it takes place inside the MinION is outlined. Then, the general structure of analysis pipelines is described, along with currently available software implemented in these pipelines and their respective strengths and weaknesses. It should be noted that nanopore sequencing is a rapidly advancing field. While some work discussed in this chapter is considered cutting-edge at the moment of writing, the reader is advised to keep the publication date of said work in mind.
In a nutshell, the underlying principle of nanopore sequencing can be explained as follows: a microscopic opening wide enough to allow single-stranded DNA to pass - the nanopore - is introduced in an insulating membrane between two compartments filled with saline solution and an electric potential is applied across it. DNA strands are then added to one compartment and allowed to diffuse toward the nanopore, where they are captured by the electric field and threaded through the pore. While a strand is passed through, the characteristic way in which the bases influence the electric current through the nanopore is measured. These measurements can then be decoded to retrieve the sequence of the DNA strand (Figure 1).
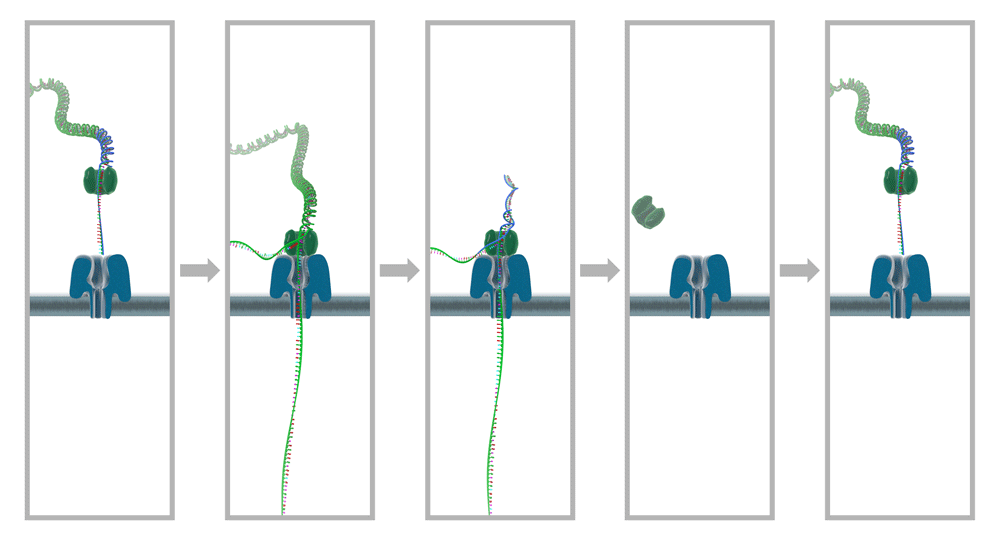
From left to right, double-stranded DNA with attached motor protein attaches to a pore protein in an insulating membrane. The applied potential pulls one strand through the pore, while the motor protein unzips the DNA in a step-wise fashion. After the DNA has been unzipped completely and one strand has passed through, the complex detaches from the pore entrance and the pore is ready to receive another strand. Image courtesy of Oxford Nanopore Technologies Ltd5.
In recent years, several key discoveries have brought reliable nanopore sequencing closer at a rapid pace. In a step-by-step exploration of the sequencing process, these discoveries will be discussed next.
Choice of pore: Biological versus solid-state Nanopore sequencing efforts are sub-categorized in two groups based on the choice of nanopore. Most current efforts implement biological nanopores, which are protein polymers derived from naturally occurring counterparts. Through genetic engineering, biological nanopores are modifiable in terms of dimensions and placement of electrical charge. These properties are also highly reproducible from one pore to the next. Functionality can be further modified by attaching compatible enzymes to the pore opening. Like their naturally occurring counterparts however, they need to be embedded in lipid bilayers, which are generally prone to disruption, particularly when exposed to varying electrical potentials. In the MinION, this was partly solved by constructing membranes out of a more stable single layer of polymers, rather than the traditional bilayer. On the other hand, solid-state nanopores are made by burning openings in a synthetic membrane using a focused electron or ion beam8. Contrary to biological nanopores, solid-state nanopores are compatible with a wide range of strong and chemically stable materials with equally diverse properties. Pores are also more easily parallelized and integrated in electrical readout circuits. A major disadvantage at the moment is the reproducibility of the pore dimensions. They also do not combine as easily with modifying enzymes. As a result, solid-state nanopores currently produce noisier and less easily interpretable signals than biological nanopores. In the following, the focus will lie on biological nanopore sequencing and the term nanopore will refer to the biological kind.
Structure and charge of the nanopore One important structural property that makes a biological pore suitable for DNA sequencing is a constriction site at which the passing strand exerts the most influence on the electrical current. The length of the constricting passage largely determines how many bases simultaneously influence the electrical current and thus the number of bases k that is “read” simultaneously at a given time. k should be kept low enough to allow recognition of a signature current for each different combination of k bases, or k-mer, and high enough to allow for some overlap between subsequent k-mers, as this benefits basecalling accuracy by allowing every base to be read k times. Modified versions of both pore proteins that have seen application in the MinION, MspA (designated R7 by ONT) and the currently used CsgG9 (designated R9, Figure 2), which have a constricted passage that allows detection of manageable k-mer lengths. For the 10Å-long constriction of the CsgG pore, basecalling models assume that five nucleotides influence the current at one given time sufficiently to discern all different nucleotide combinations, and thus 5-mers are called.
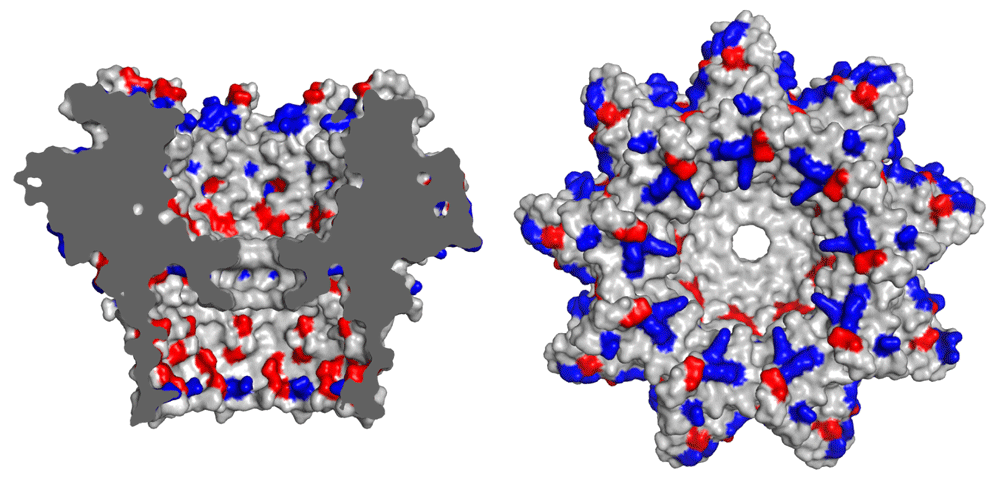
Positive and negative residues are colored blue and red, respectively. Image generated by the authors using PyMOL v1.7.0.0. PDB ID: 4UV39.
For sequencing to commence, a DNA strand first needs to diffuse towards one side of the pore, referred to as the cis-side, where it is captured by the electric field resulting from the applied potential. It is then threaded through the pore and extruded at the other end, called the trans-side.
Two forces should be considered. First and most importantly, the electrophoretic force induced by a positive electric potential applied at the trans-side attracts the negatively charged DNA and pulls it in. As negative particles leave the cis-side and positive particles simultaneously move in the opposite direction, a positively charged zone forms around the cis entrance of the pore, strengthening attraction of DNA strands. Secondly, strand translocation is influenced by the electro-osmotic flow (EOF), the force induced by the net water and ion flow through the pore. While a DNA strand is in the pore, the EOF normally opposes the direction of the electrophoretic force and thus of translocation; however this effect is relatively minor.
Through iterative optimization of internal architecture, it was found that positive internal surface charges are important for efficient DNA capture10,11, while base recognition was found to improve with bulky or hydrophobic amino acid side chains placed at the constriction site, as these direct ion flow toward the DNA strand12. Although the structure of the modified CsgG pore9 implemented in the current MinION flow cells (R9-type) has not been publicly released by ONT, modifications to these properties have likely been made.
Processive control It should be noted that the processive speed of the strand is too high without any further modifications (between 2·106 and 10·106 bases/s in wild-type MspA)10. Currently, the most successful way to exert control over the speed has proven to be the addition of a motor protein, such as phi29 DNA polymerase (DNAP)13 or a helicase14. In a preparatory step, a poly-T or "leader" adapter is attached to the double-stranded DNA. The motor protein attaches to this adapter, but due to specialized bases in the adapter sequence (left unspecified by ONT), it cannot unzip it at this stage (see community technical document on this subject). Once the complex is adjacent to the cis-side of the pore, the leader adapter previously blocking the motor protein is released, presumably due to the force exerted on the strand as demonstrated by15 and described in Heron et al., 201616. The DNA is then fed through the pore by the motor protein, where it can now be "read" at a reasonably regular pace. Presumably, a modified helicase is currently used as motor protein in the MinION14. The latest release of this motor protein at the moment of writing (dubbed E8) maintains an average throughput speed of 450 bases/s.
Reading the DNA strand During a MinION sequencing run, the potential over the membrane is kept stable, while the electrical current (in the pA-range) is sampled at a frequency in the kHz range (Figure 3). This signal is characteristic for the subsequent bases moving through the pore and will ultimately serve as the basis for basecalling. As the amount of electrolyte is increasingly depleted during the run, the applied potential (typically starting at -180mV) is further decreased by 5 mV per two hours of runtime and increased by 5mV when the MinION switches to another set of wells filled with fresher buffer (see next section).
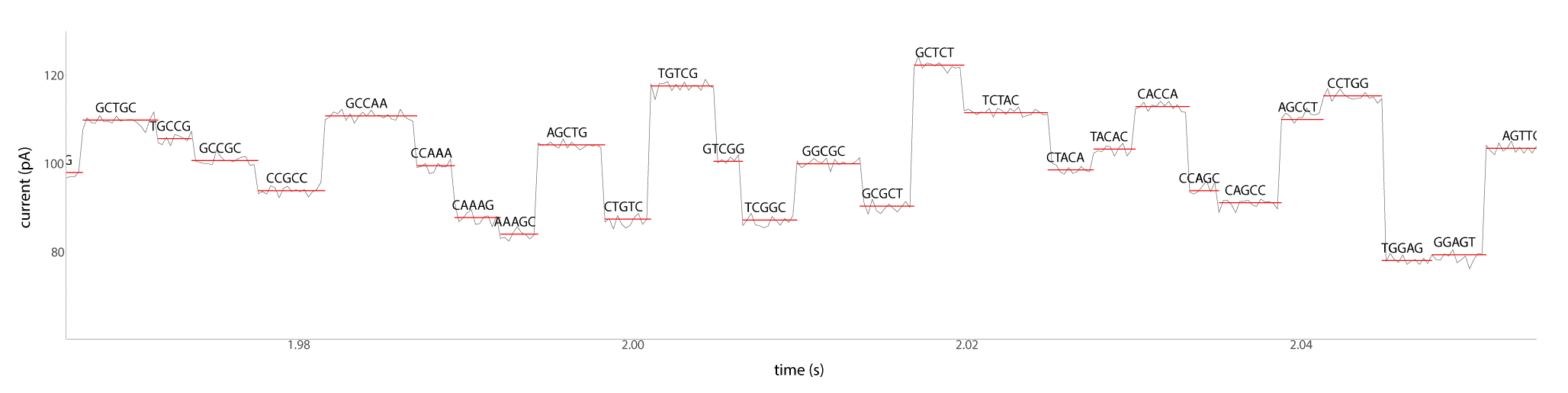
The data used in the construction of this figure is mock data.
While the MinION can read the first strand of a dsDNA-stretch that is threaded through the pore - by definition, the template strand - and discard the complementary strand, it is possible to instead read the complementary strand immediately after the template, thus performing a second read of the same stretch (Figure 4). Combining reads of both strands has been shown to increase sequencing accuracy significantly5. So far, ONT has provided two ways of doing so; 2D-sequencing and its successor 1D2-sequencing (versus 1D sequencing if only the template strand is read). To enable 2D-reads, an abasic hairpin sequence (i.e. a bare backbone of DNA) is attached to the dsDNA, connecting the 3’-end of the template and the 5’-end of its complement. After the template strand has been read, the hairpin structure is threaded through the pore, recognizable due to its abasic nature, followed by the complement strand. The mirroring reads are then decoded jointly so that any sequencing errors may be corrected. It should be noted however, that decoding 2D-reads comes at a high computational cost. Furthermore, the exact start and end of the hairpin is not always clear from the signal. The hairpin may also ligate strands in other ways than the intended one; for example, multiple reads may be linked together by hairpins into chimeric reads17. Lastly, the read of the complement strand is often less accurate and threaded through the pore at lower speed5, reportedly due to structural changes in the strand caused by the template and complement rezipping while the complement strand is still passing through the pore. In May of 2017, 1D2-read chemistry was introduced as a replacement of 2D-read chemistry. 1D2 chemistry does not join the two strands, but rather attaches an adapter to the complement strand, allowing it to attach to the membrane while the template strand is read. Shortly after the template strand has completely left the pore, the complement strand is pulled in and sequenced. Without the hairpin connecting the strands, recognition of its start and end is no longer required and, as the strands are effectively read as two separate reads, no drop in read quality is observed for the complement strand.
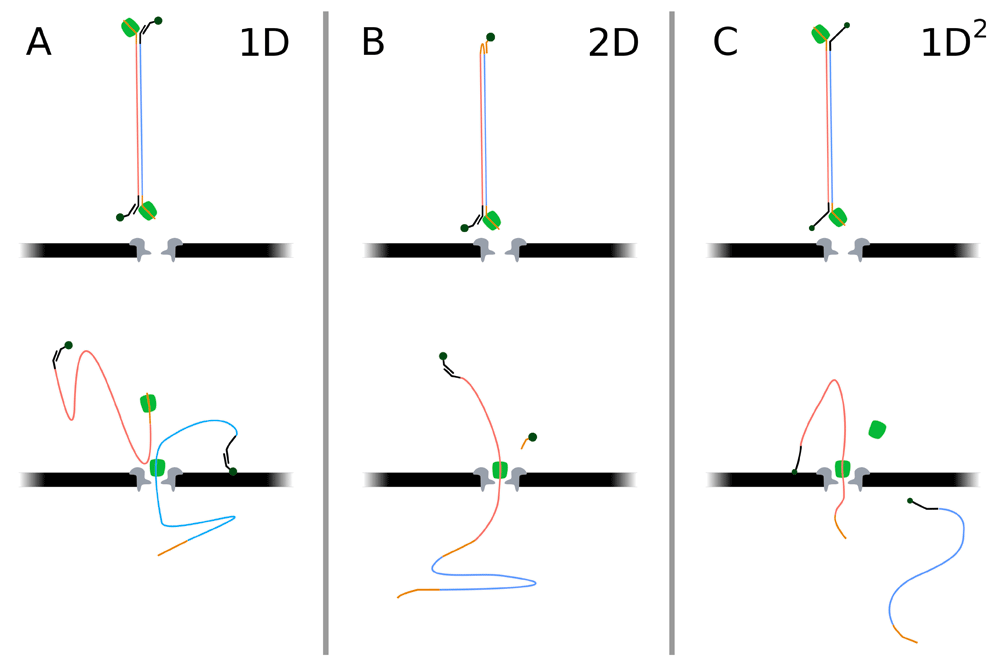
(A) When using 1D chemistry, only the template strand (blue) is threaded by the motor protein (green) and read. The complement strand (red) is discarded at the cis side of the pore. The tethers (dark-green) allow for selection of properly ligated complexes during sample preparation and attach to the membrane to increase the availability of strands near pores during sequencing. (B) The now-deprecated 2D chemistry connected template and complement strand using a hairpin, thus allowing sequencing of the complement strand immediately after the template strand. An additional tether that attached to the hairpin allowed for selection of correctly ligated strands during sample preparation. (C) 1D2 chemistry, the successor of 2D, also allows sequencing of both strands, but rather than attaching the two, the complement strand is tethered to the membrane while the template is sequenced. After the template strand is threaded through, the complement strand is drawn in and the tether is pulled loose. Based on Jain et al., 201518 by permission from Macmillan Publishers Ltd: Nature Methods, copyright(2015), the ONT kit content description, and ONT’s technical update of March 2017.
Channel parallelization Lastly, throughput can be greatly increased by reading the signal from multiple pores in parallel. The current generation of the MinION’s disposable cartridges, called flow cells, can read the signal of up to 512 pores in parallel (Figure 5). The flow cell is equipped with 2048 wells, which are connected in groups of four to multiplexers (MUXs), the switches that control which of the four cells per group is controlled and read out by the circuits. During the initial platform quality check, DNA strands (of unreleased source and sequence), present in the buffer with which the flow cells are shipped, is sequenced to discern wells containing precisely one correctly inserted pore from wells in which correct pore insertion has failed (e.g. no or multiple pores were inserted; see ONT platform quality check protocol). The latter scenario may occur, as the insertion of pores is a stochastic process. In a second quality check, the MUX scan, each MUX chooses three wells in order of signal quality and begins readout in the best-quality well. As well quality is expected to decline during the run, the standard protocol switches to the second-best quality pore after eight hours, and the third-best quality after another eight hours. This way, the best and most output is expected in the first part of the run. While a run using a group of wells is in progress, the circuits connected to the MUXs regulate the current in each selected well individually. This also allows expelling of eventual blockades from a pore, by temporarily reversing the current in the affected well while the rest of the wells continue to function normally.
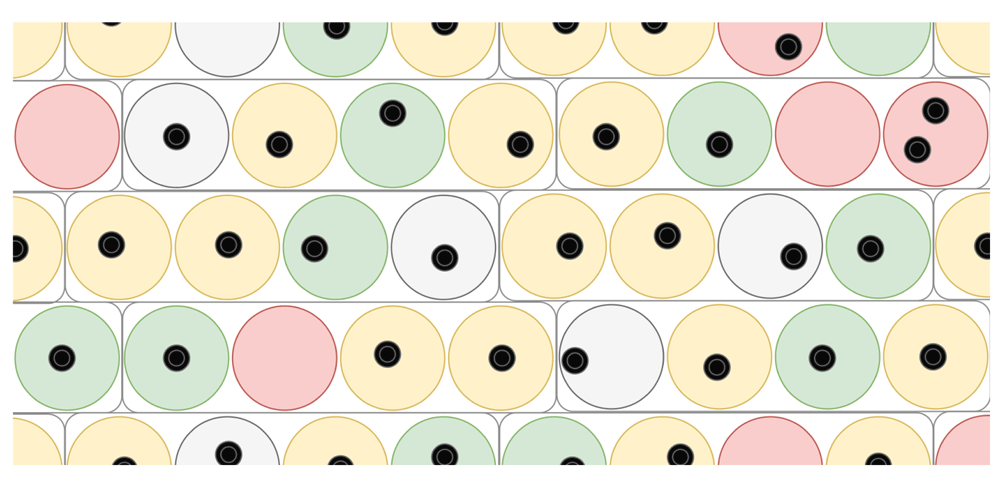
Large circles denote wells in the grid, small black circles denote inserted nanopores. Each group of four wells is controlled by a multiplexer (MUX). During an initial quality check, wells that are unusable due to erroneous pore insertion are marked unusable (red circles). Right before sequencing, the wells are tested a second time and three wells per MUX are ranked on signal quality (if possible). Sequencing of the sample will then commence, starting read-out from the best-performing well (green) and switching to second and third best (yellow) after eight hours each.
Following the process in section 1, a current signal is obtained that needs to be translated into the corresponding DNA sequence. Pipelines used for this purpose generally subdivide the signal into discrete stretches corresponding to a particular set of k bases, called events, and then attempt to find the most likely set of bases for each event6,7. Ideally, the processive control exerted by the motor protein causes the strand to move one base further through the pore between each event transition, such that each base in the k-mer is read k times. Due to irregularity in the motor protein functioning and noise in the signal, this is not always the case. Three transition scenarios may be observed. (I) If the strand progresses exactly one base further from one event to the next, this is called a "step". Alternatively, (II) the next event may denote a k-mer located one or multiple bases further upstream, which is referred to as a "skip", or (III) the segmentation process may incorrectly split up an event in two events that correspond to the same k-mer, which is called a "stay". Steps, skips and stays may be identified by comparing the bases in the read k-mers; however, this proves more difficult in short tandem repeat regions, as subsequent events would look the same for multiple transition scenarios. More specifically, in a homopolymer stretch of length k or longer, the difference between skips, steps and stays may be unclear. Similarly, repeats of m bases may cause problems in discerning skips of (multiples of) m bases and stays. Such ambiguities have been shown to be a major cause of deletions in current basecallers6,18–20.
To assess the quality of basecalling performance, a 3.6 kbase calibration strand derived from the Lambda genome may be added to the sample. The software controlling the MinION (MinKNOW) automatically detects reads derived from the Lambda genome and separates those from the sample reads. Some software tools use these strands to parameterize basecall correction algorithms21.
Once reads have been basecalled, they may serve several purposes. If SGS reads are available, then those may then be mapped to the MinION reads to correct sequencing errors pre-assembly22, or to create large low-error contigs. The latter goal may be achieved either by using MinION reads as scaffolds to properly align short reads23,24 or by creating short accurate seed regions from short reads, the gaps between which are then bridged by MinION reads20. Both approaches were shown to result in accurate and highly continuous de novo assemblies and identification of repeats that were collapsed in SGS-only assemblies20,22. In MinION-only assembly pipelines, overlaps between the basecalled sequences are sought to create initial assemblies. Currently, the error-prone nature of MinION reads necessitates a post-assembly error correction ("polishing"), in which raw reads are mapped again to the assemblies until a better consensus is reached25. Below, the different tools MinION users have at their disposal to fill in the steps in this pipeline are discussed, along with their strengths and weaknesses.
It should be noted that the presented list of tools is not exhaustive; the focus of this section lies on tools that can be used in de novo MinION sequencing, without the need for other sequencing methods. Furthermore, only tools that provide a full solution to their respective step in the assembly pipeline are reported here. Notably, stand-alone read mapping tools exist, which may be used in combination with the assembly tools described in this section; however, detailed descriptions are omitted for the sake of brevity. Readers with an interest in these alternatives may want to look into GMAP26, Graphmap27 and MinHash alignment process (MHAP)28. As current assemblers either include their own error correction module29 or work with uncorrected reads20,30–32, stand-alone pre-assembly error correction tools are excluded as well.
Before basecalling takes place, current basecalling tools require the continuous signal to be divided into discrete sections, called events, each supposedly representing one combination of bases. Furthermore, the poly-T lead preceding the sequence needs to be removed, and, if 2D chemistry is used, the signal derived from the template strand needs to be separated from that of the hairpin and the complement strand. The latter process is commonly referred to as segmentation.
For locally basecalled reads, both segmentation and subsequent event detection are performed by the MinKNOW software provided by ONT. The software identifies the hairpin by a characteristic double current spike, while a long poly-T signal denotes the lead. For event detection, MinKNOW was reported to calculate a simple t-statistic between sliding adjacent windows of set size. Peaks in the t-statistic above a certain threshold are then assumed to signify the border between two segments. Optionally, the raw current signal can also be reported for further analysis.
Several dedicated basecalling tools are already available to MinION users. In this section, the underlying principles and implementation of these tools are explored, along with their reported subsequent strengths and weaknesses. It should be noted that an all-inclusive fair comparison, in which all basecallers are optimized for the latest MinION chemistry and then run on the same dataset, is currently lacking. Any comparison in this chapter is therefore based on accuracy reports made by the authors of the open-source basecallers in their publications (i.e. between the Metrichor basecallers and their own).
Metrichor basecallers Metrichor, a spin-off company of ONT and its main developer of proprietary analysis software, maintains a range of basecallers that have remained the go-to option for most MinION users. Initially, the Metrichor basecallers relied on hidden Markov models (HMM) to find the biological sequence underlying the segmented signal. As of early 2016, this model was replaced by a more accurate recurrent neural network (RNN)-implementation. Currently several RNN-based versions exist under different names; Albacore, Nanonet and the MinKNOW integrated basecaller. Albacore and the MinKNOW version are stable versions intended for regular MinION users. As of Albacore v1.0.1 and MinKNOW v1.6.11, both were reported to improve homopolymer calling through the implementation of a transducer, a feature that was transferred from the Scrappie basecaller (see below). Nanonet is an unsupported version under continuous development and makes use of the CURRENNT library to implement its RNN33. A cloud-based version was previously integrated in the EPI2ME platform, but this service has been discontinued. Due to the proprietary nature of the software, the source code of Albacore and the MinKNOW basecaller is currently only open to developer users, while that of Nanonet is openly available. The community manual describes the general procedure followed by all Metrichor basecallers as follows. Long reads are broken up into partially overlapping stretches, referred to as chunks. This keeps the Viterbi-decoding process, required after the RNN has determined k-mer probabilities, tractable. The RNN then assigns a likelihood for every possible five-mer to each event in the chunks.
Metrichor basecallers were reported to use a bidirectional long-short term memory (BLSTM)-RNN. This architecture allows the RNN to overcome the vanishing gradient problem, encountered often during the training of deep neural networks, and the usage of multiple previous events in the sequenceii.
The RNN assigns probabilities for each k-mer to each event, which are used to estimate basecalling quality scores (i.e. q-scores). Five-mers that would entail a forbidden move, considering the likelihood of surrounding k-mers, are removed at this point. The maximum likelihood sequence is then extracted from the k-mer likelihoods through Viterbi decoding. Finally, basecalled chunks are merged again.
The accuracy of the Metrichor basecallers is currently considered to be the highest of all available basecallers. Before the switch to RNNs, an international performance analysis estimated the accuracy at 88% for 2D basecalling5. David et al. reported an accuracy of approximately 68%6 for 1D basecalling on human data, while Boza et al. reported 77% for 1D and 87% for 2D on Escherichia coli and Klebsiella pneumoniae genomes. After moving to an RNN-implementation (as well as numerous improvements in used chemistry and hardware) this percentage has risen to over 95% (see here for a publicly available dataset supporting this), even before the mentioned correction of homopolymer regions in Albacore and MinKNOW was applied.
Scrappie A basecaller by Metrichor and platform for ongoing development, Scrappie was the first basecaller reported to specifically address homopolymer basecalling, through the implementation of what is referred to as a transducer. It was written in C and its source code is publicly available. In previous MinION sequencing efforts, correctly determining the length of homopolymers proved challenging, as the separation between states in a homopolymer stretch was not clear20,21,35. Scrappie can process the raw current signal, rather than the event-detected data.
As an initial performance assessment, an early-development version of Scrappie was run in parallel to other Metrichor basecallers on reads of the human chromosome 20. It was found that Scrappie indeed called homopolymer stretches more accurately than the EPI2ME basecaller and Nanonet; homopolymeric stretches of up to 16 bases were called accurately (although a slight over estimation of repeat units was seen between 5 and 15 bases), after which the accuracy fell. In contrast, the EPI2ME basecaller and Nanonet failed to call stretches longer than five bases. The effect was also shown in an analysis of 5-mer counts; of the tested basecallers, Scrappie stayed closest to the reference genome and did not under-represent homopolymeric 5-mers3. Although these results are promising, a thorough assessment of Scrappie in a later stage of development is required to evaluate the true potential of the transducer-based homopolymer calling.
Nanocall Nanocall6 was the first open-source basecaller for the MinION offered as an alternative to the proprietary Metrichor software. It was written in C++. Nanocall accepts the segmented signal from MinKNOW and assigns k-mers to the events using a HMM.
An HMM assumes that, in a sequence of events, the probability of the next event being of a certain nature - its state - is dependent only on the current state. This makes sense for the basecalling of subsequent k-mers, as the prefix of the next k-mer that is called should be the suffix of the previous. The "hidden" property of HMMs refers to the fact that the sequence of states - the path - is not known. Instead, it has to be inferred with some probability from a derived signal. In the case of a basecaller, this signal consists of electrical current levels which are derived of a sequence of k-mers. The goal of the HMM is to find the path that is most likely to have emitted the given signal (for a more thorough introduction to HMMs, see 36).
As described by David et al.6, the HMM underlying Nanocall takes stay, step and skip scenarios into account at each event transition (Figure 6). Although skips of more than one base may occur in a sequence, Nanocall only considers one-base skips to cut down on computational requirements, thus reducing the number of neighboring states to 21 (i.e. stay, four different steps and sixteen different one-base skips). During an initial training phase, state transition probabilities for stays and skips of any number of bases, pstay and pskip respectively, are estimated using the Baum-Welch algorithm (pstep is derived as 1 − pstay − pskip)37. The transition probability for exactly one base pskip1 is then derived as pskip1 = pskip /(1 + pskip). Note that all sixteen one-base skips have the same transition probability (i.e. · pskip1), as do the four step scenarios ( · pstep). To calculate emission probabilities, Nanocall relies on ONT’s pore models, which provide parameters for current level distributions per k-mer, which can be fitted to individual reads. The most probable sequence is obtained through Viterbi decoding.
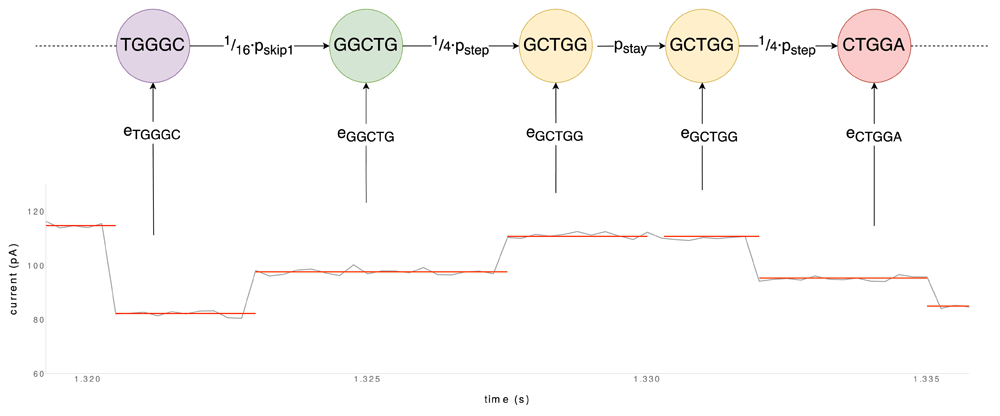
The path is chosen by maximizing the product of transition probabilities pstay, pstep and pskip1 (modified by fractions in the case of skips and steps, as there are multiple states to which these transitions could lead) and emission probabilities of k-mers. The data used in the construction of this figure is mock data.
Performance of Nanocall at the moment of its publication, in March of 2016, was reportedly on par with that of 1D analysis by the Metrichor basecallers, or around 68% accuracy6. However, unlike the Metrichor basecallers, Nanocall does not support 2D analysis, which allowed for a much higher accuracy in the comparison by David et al. (around 85%)6. Since the Metrichor basecallers switched to a recurrent neural network-approach, its 1D accuracy proved significantly higher than that of Nanocall as well. Moreover, Nanocall is incapable of calling homopolymer stretches longer than its k-mer length, while current Metrichor basecallers are able to do so. Therefore, as the authors currently state on the Nanocall Github, it should be seen as a platform for testing out novel ideas, rather than a prime choice for basecalling MinION data.
DeepNano Before Metrichor made its own switch from HMM- to RNN-basecalling, the open-source basecaller DeepNano7 already implemented a form of RNN-basecalling, booking a significant improvement in accuracy with respect to the then-current Metrichor version (late 2015). DeepNano was written in Python, using the Theano library38.
Rather than LSTM-nodes, as currently used in Metrichor basecallers, DeepNano implements gated recurrent units (GRUs)39. Although the design of GRUs is simpler than that of LSTM units, performance assessments show that GRUs are able to achieve similar or slightly better accuracy in different learning tasks40,41.
DeepNano’s RNN was trained separately for (hairpin-connected) 1D basecalling and true 2D basecalling. As the exact alignment to the reference may be uncertain, the algorithm alternately trains the RNN weights for 100 iterations and then re-aligns the reads to the reference. At the moment of its release in March of 2016, DeepNano was reported by its authors to make more accurate calls than the Metrichor agent for both 1D calling (approximately 70% versus 77%) and 2D reads (approximately 88% versus 87%). However, as the Metrichor agent has since then switched to an RNN-approach as well, these comparisons are outdated.
In the assembly stage, basecalled sequences are combined to reconstruct continuous parts of the genome as accurately and completely as possible. Assembly of nanopore reads requires a different approach than that of SGS reads; as nanopore reads are longer, finding a correct overlap should be easier, yet they are more error-prone, which increases the uncertainty of overlaps. Because of these differences, a return of interest in overlap layout consensus (OLC) algorithms - which were at the peak of their popularity in the era of Sanger sequencing - is seen. De-Bruijn graph (DBG) assemblers, the more popular choice for SGS reads, were reported to return lower quality assemblies of nanopore reads than OLC-based methods, but proved faster in some cases42. A more detailed comparison between OLC and DBG assemblers, along with implementations of both approaches is discussed in this section. Software using traditional greedy extension algorithms (e.g. SSAKE) was found to perform decidedly less well in a de novo assembly setting, both in terms of assembly quality and required computational resources42, and is therefore omitted here.
PBcR & Canu Originally developed for the first human genome draft, the Celera assembly pipeline43 and its extensions [Koren et al., 201729, Miller et al., 200844, Zimin, 201345, i.a.] have remained a popular choice in a growing landscape of OLC assemblers. Briefly, the Celera assembler uses read overlaps to find contigs of which the structure can unambiguously be derived from overlap information, referred to as unitigs. It then separates repetitive unitigs from unique ones and attempts to orient the unique unitigs with respect to each-other. Where possible, gaps between unique unitigs are filled with repetitive elements. As a high read error rate is detrimental to the quality of the assembly46, two different modifications to the pipeline are available. The PacBio corrected Reads (PBcR) algorithm, originally developed for the correction of PacBio reads suffering from similar error rates, uses accurate short reads mapped with high confidence to the long reads to correct errors. The assembly then proceeds as is usually done by Celera47. PBcR’s successor, Canu29, provides a solution that is more integrated with Celera and does not require short accurate reads. The pipeline includes three stages; correction, trimming and assembly. Overlaps are found using the efficient MHAP28, which hashes k-mers using different hash functions and for each hash function stores the smallest integer to which a k-mer of the sequence is hashed. Comparing the hashed k-mers per read results in initial overlap hits, which are then used to perform error correction by consensus seeking. By selecting overlaps for correction on quality, but limiting the number of overlaps a read can contribute to, Canu attempts to prevent masking of true repeat variants. Shorter reads are used at this stage to improve accuracy of longer reads. In the trimming step, overlaps are recalculated to locate and filter out regions of low coverage and high error. Reads are overlapped two more times to correct specific types of errors (i.e. missed hairpin sections/adapters, chimeric reads) and adjust error rate per overlap, before the actual assembly phase starts. With adjustments to account for erroneous alignments and residual errors, assembly essentially follows the same procedure as CABOG, another Celera-based pipeline44.
Due to its thorough yet relatively efficient correction steps, Canu is significantly more accurate than both its predecessor Celera/PBcR and Minimap/Miniasm25. In a benchmarking effort on Enterobacter kobei reads, it produced an assembly of higher contiguity, with fewer indels and mismatches. This is in line with the accuracy assessment by its authors29.
Minimap & Miniasm In terms of speed and computational efficiency, the OLC-based pipeline consisting of Minimap and Miniasm32 has a definite advantage over other existing tools25. This efficiency was reached through the omission of the consensus step and the use of minimizers. Much like the output of the MinHash algorithm used by Canu29, a minimizer is a memory-efficient hashed representation of a sequence. Minimap computes the set of minimizers of a sequence, the “sketch”, by finding the k-mers represented by the smallest hash value within a certain window size of each position of the sequence (Figure 7). The inverse of each k-mer is also considered. Decreasing the window size will increase the returned number of minimizers and allow for more accurate alignment at the cost of increased computational requirements. Minimap then performs all-versus-all mapping by identifying hits between minimizers of different sequences. The found overlaps are passed on to Miniasm, which constructs an assembly graph. First, artefacts are trimmed from each read by removing all sequences outside the longest stretch with more than 3 times coverage. Then reads contained within other reads are removed and small bubbles, less than 50 kb in length, are popped (i.e. a consensus is taken in cases where paths split and later join up again). Finally, sequences can be extracted from stretches of the graph without multi-edges to form unitigs. The error rate at this point is practically the same as that in the raw reads, emphasizing that correct basecalling is essential for the eventual quality of the assembly. The graphical fragment assembly (GFA) output format of Miniasm conveniently allows both graphing of the uncorrected assembly and addition of consensus error correction tools, such as Nanopolish or Racon, to the pipeline, though the latter increases walltime and computational cost severely25.
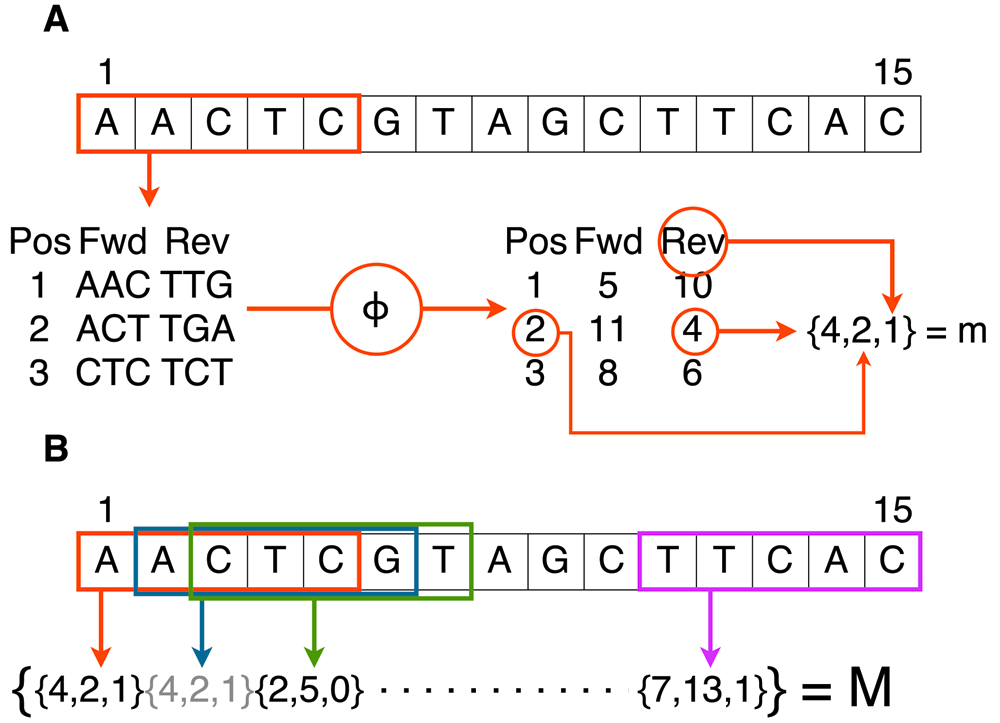
(A) To extract one minimizer m, all k-mers and their complements are listed and hashed to a distinct integer using hash function φ. The smallest hash value is then stored with its starting position and a binary value denoting the strand on which it was found (0 for forward and 1 for reverse). If a multiple of the smallest hash value is found within a window, all are stored. (B) The minimizers of all windows in the read are collected and stored as the sketch, M. As M is a set, double minimizers are removed.
In March of 2016, the authors of Minimap and Miniasm reported assembly of MinION reads of an E. coli genome in a single contig. In May of the same year, Judge et al. assembled an Enterobacter kobei genome in 16 contigs with an N50 of 662 kbase in two minutes, while the next fastest assembler (Canu) took two hours. However their benchmark showed that the omission of an error correction step caused the eventual assembly quality of E. kobei to be too low to properly assess by the QUAST analysis tool25. The inclusion of the Nanopolish consensus correction tool after Miniasm improved the quality of the assembly enough to perform a reliable quality analysis. However, the pipeline was still under-performing compared to other tools.
TULIP As more reads are required to cover larger genomes, and as the time required for all-vs-all overlapping increases quadratically with an increasing number of reads, it follows that the overlap step of OLC assemblers may take unfeasibly long for very large genomes. To tackle this issue, The Uncorrected Long read Integration Process (TULIP) takes a different approach to read overlapping20. Instead of all-vs-all alignment, short seed sequences are selected, which the assembler then attempts to align with long reads. This drastically cuts down the overlapping complexity and makes efficient use of long reads to cover long stretches of the genome between the seed regions. The resulting graph represents seeds as vertices and the connecting reads as edges. In a graph cleaning step, vertices with multiple in- or outgoing edges are revisited. Spurious and superfluous edges are removed aggressively, thus producing a linear graph. Note that, as the name implies, TULIP does not perform basecalling error correction.
The success of assembly using TULIP highly depends on proper seed selection. To avoid spurious connections between reads, the seeds need to be sufficiently unique in the genome and contain few sequencing errors. If available, SGS reads may be used to construct seeds, although with the increasing accuracy of MinION reads, the ends of long reads may be used as well. Apart from cutting out the need for SGS methods, the latter approach has the added advantage that pairs of seeds are connected by at least one long read. Furthermore, as TULIP is not able to assemble regions in which the gap between seeds is larger than the read length, a proper seed density over the entire genome is required. If a marker map is available for the genome, this information can be used to control the distribution of seeds in the selection process.
As a first demonstration of TULIP’s efficiency, Jansen et al. assembled the genome of the European eel Anguilla anguilla (approximately 850Mbp) with 18x coverage in three hours (excluding sequence polishing), requiring only 4.4GB of RAM and four threads20. The resulting assembly was more continuous than the SGS-based reference genome. As was the case with Minimap/Miniasm however, the current quality of MinION reads combined with the lack of an error correction step necessitates post-assembly correction. The authors further showed that missed seed alignments were the most commonly encountered issue during graph simplification, followed by tangled alignments due to repetitive seeds and spurious alignments. The seeds, constructed from short SGS reads, only underwent selection by uniqueness, which did not lead to an equal distribution over the entire genome; however, density remained high enough for successful assembly. The authors noted that assembly using the tips of MinION reads as seeds proved successful for Escherichia coli genomes, but this has not been attempted for larger genomes yet (personal communication, May 1, 2017).
HINGE Although long reads provide a definite edge when attempting to resolve repeat regions, issues may still occur if not all individual repeats are spanned by at least one whole read. In such cases, HINGE may provide a solution. Rather than attempting to resolve frayed rope structures in the assembly graph afterwards, HINGE preprocesses the reads to separate repeat regions that are entirely spanned by a read (and are thus more easily resolvable) from those that are not, and collapses the latter beforehand30.
First, HINGE attempts to identify reads that wholly or partly overlap a repeat region. It does so by performing all-vs-all alignment and then selecting those reads of which a stretch aligns to a proportionally larger number of other reads than the rest of the read. The intuition behind this is that reads from all copies of a repeat region existing in the genome align to each other, thus causing a characteristic abrupt increase in alignments for reads that overlap these repeat regions. Repeat regions covered entirely by at least one read can be easily resolved and are omitted from the following procedure. Of the reads lining the same repeat region, the reads that extend furthest into the repeat region (regardless of the location of the actual copy), are designated “hinges”. In the subsequent greedy extension of the hinges, the path will split at the hinge regions. Like Miniasm, HINGE outputs its assembly in the form of a graph. As its authors show, this is particularly useful for circular genomes.
HINGE provides an elegant solution to long repeat resolution, by separating resolvable regions from unresolvable ones on forehand. Its authors compared HINGE to Miniasm on PacBio reads of 997 circular bacterial genomes and found that overall, HINGE produced a completed genome in more cases than Miniasm could30. Whether the precaution taken by HINGE is necessary is dependent on the genome under consideration and the used reads; if the genome is known to contain repeats longer than most of the reads, the described approach would be justified.
ABruijn While more traditional DBG assemblers performed relatively less well than OLC assemblers on assembling long error-prone reads42, the adapted approach taken by the ABruijn assembler has shown more promise31. To account for the high error rate, ABruijn filters all k-mers occurring in the reads by their frequency; if a k-mer occurs relatively few times, it is assumed that it contains basecalling errors and it is removed. Then, k-mers are fused into so-called “solid strings”, sequences that contain no other occurring sequences as substring. The ABruijn graph is then drawn by representing solid strings as vertices and connecting them where connections exist in the reads. The edges are weighted by the number of positions between the first bases of the connected solid strings. The assembler consults the weights in this graph to quickly identify overlaps between reads, allowing to select on a minimum overlap length and maximum overhang length. The assembly graph is constructed by starting with the graph for an arbitrary read and continuously extending it by overlapping it with other reads. ABruijn also includes an error correction routine, during which a best consensus between reads is found by identifying low-error stretches and, in between those stretches, choosing the consensus sequence that maximizes the likelihood of the read sequences.
Unfortunately, the authors of ABruijn evaluated their assembler only on E. coli MinION reads of an older and more error-prone chemistry (R7.3), and compared their assembly to that of a Nanocorrect and Celera pipeline, which is currently considered obsolete. In this comparison ABruijn performs better, producing an error rate of 1.1% versus 1.5% for Nanocorrect/Celera, with the majority of errors being deletions. How ABruijn stacks up against currently popular assemblers optimized for MinION data on reads produced with up-to-date chemistries is unclear. The more extensive assessment on PacBio reads provided by the authors shows a dramatic decrease in error rate in the ABruijn assembly in comparison to Canu’s, but requiring comparable running time.
A number of tools attempt to improve assemblies by remapping reads to the assembly and adapting the assembly to increase local resemblance to the reads. These tools may be essential to use after assemblers that do not include a consensus step themselves, such as Minimap/Miniasm and SMARTdeNovo, but have also frequently been used to polish assemblies produced by the other assemblers.
Nanopolish Nanopolish attempts to find an optimal consensus between an assembly and the raw current signal output by the MinION, by iteratively proposing and evaluating small adaptations to the assembly based on the original reads35. The proposal mechanism for adaptations works in two steps. First, reads are aligned to the assembly and the resulting multiple alignment is divided in 50 bp subsequences of the assembly. For each read aligning partly or fully to a subsequence, sections in which 5-mers perfectly align to the assembly are detected. The consensus sequence between each pair of aligning sections is replaced by the aligned read subsequence, creating an initial set of alternative candidate sequences. In the second step, this set is further extended by proposing every possible one-base deletion, insertion and substitution in the previously generated candidate sequences. Of this set, the sequence maximizing the likelihood of observing the raw signal is picked. This process allows Nanopolish to explore a decent number of likely modifications, while remaining computationally tractable.
Polishing an assembly with Nanopolish was found to improve assembly quality, regardless of the assembly tool used. One study on E. coli sequencing data reported that identity to the reference genome rose from 89% to 99% when Nanopolish was applied after Minimap/Miniasm, while improvement after Canu was more modest (98.2% to 99.6%)48. Although it addresses all types of errors, a large part of the increase in accuracy is reached due to the correction of homopolymeric stretches. Despite its efficient searching heuristic of block replacement and mutation, running Nanopolish remains a time-consuming step; on the error-prone assembly produced by the otherwise quick Minimap/Miniasm pipeline, it may take up to a few extra days of CPU time to finish polishing25,48.
Racon Racon corrects MinION assemblies by finding a consensus sequence between reads and the assembly through the construction of partial order alignment (POA) graphs. After alignment of the reads by a mapper of choice (e.g. Minimap or Graphmap), Racon segments the sequence and finds the best alignment between a POA graph of the reads and the assembly. By default, the alignment is performed using the Needleman-Wunsch algorithm, which can align sequence and POA graph with little adaptation (Figure 8). The alignment process is sped up by parallelization. Racon was reported by its authors to be two orders of magnitude faster than the popular (yet currently deprecated) Nanocorrect after assembly of an E. coli genome by Miniasm, albeit not quite as good at diminishing the error rate (to 1.31% versus 0.62% for Nanocorrect). Compared to consensus steps in Falcon49 and Canu29 on that same assembly, Racon remains an order of magnitude faster while producing similar error rates. A closer look at the error rate reveals that the majority of errors are indels, validating the need for homopolymer and tandem repeat corrections even in a pipeline containing a polishing step with Racon. Finally, the total genome size estimate following application of Racon was closer to the reference genome size than the estimates of Canu, Falcon and Nanocorrect.
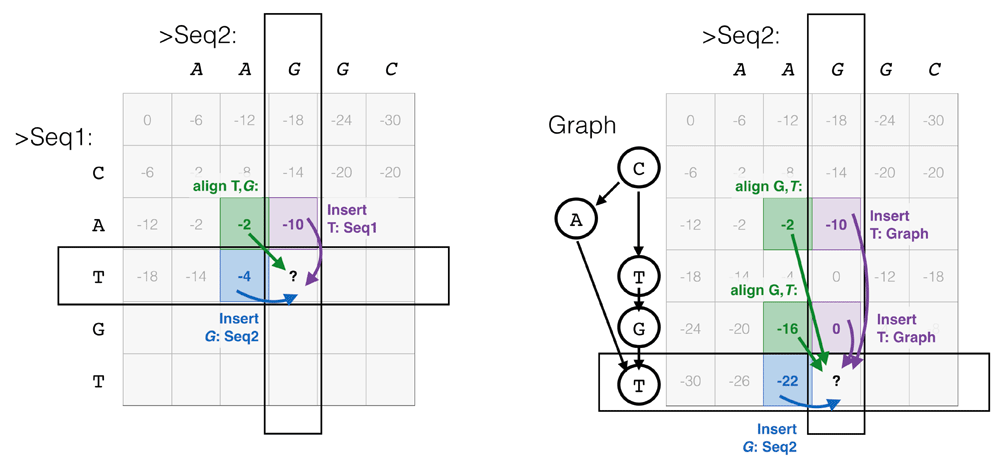
In the latter case, the graph dictates which steps in the matrix are allowed. Taken from Jonathan Dursi’s blog on POA multiple alignment, with permission.
Nanopore sequencing is a promising new venue in biology research. Inexpensive, small, capable of producing long reads and freed from the need for nucleotide labeling or amplification, it is conceivable that the MinION will make cost-effective, fast and portable de novo whole genome sequencing of even complex genomes possible in the future. In this review, an attempt was made to give an updated overview of the progress in this field, focusing in particular on de novo whole genome sequencing.
Available basecaller tools have been improving rapidly in accuracy. A notable recent improvement was made through the application of RNNs. For the next step in a typical sequencing routine, assembly, OLC-assemblers are currently considered the best option for accurate nanopore-based assembly. The choice of assembler should be adapted to the characteristics of the genome and the priorities of the user. Canu is a complete and accurate solution, while maintaining reasonable CPU times. However, for an assembler without included error correction, Minimap/Miniasm is able to produce a decent assembly and, without any post-assembly correction, is the fastest option available on smaller genomes. For large, complex genomes, TULIP may be the more tractable alternative. Lastly, consensus error correction tools are currently highly useful. Notably, Racon provides a computationally more attractive alternative to the often-used Nanopolish.
Currently, the most prominent obstacle for de novo sequencing using the MinION is the high error rate of the reads. Improving basecalling accuracy would not only improve assembly quality in a direct manner, but may also allow more computationally efficient assembly.
The active research community surrounding the MinION has booked great progress in both the development of new applications and improvements on accuracy of existing ones. ONT also continuously works on improvements for both its hardware and software platforms, and regularly updates its users on this.
Although these updates often entail welcome new features or some form of accuracy improvement, it should be noted that this policy has also lead to some difficulties. Developers may not be able to keep pace with ONT when evaluating, updating or calibrating their tools, and users may not always know which tool is suited best to their data and needs. As a result, most published studies, including tool benchmarking efforts, were conducted using older or multiple chemistries. Although such growing pains are to be expected for a novel fast-developing field of research, the MinION’s current state of development may allow for some increase in stability, thus giving the user community the time for proper evaluation.
iEstimate based on a purchase of 24 flowcells and a 1D/1D2 sequencing kit, 18th of May 2017
iiA thorough discussion of RNN architectures and their respective properties is outside the scope of this article. Interested readers are referred to 34 for an introduction to the subject.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)