Keywords
chemical space, data visualization, epigenetics, principal components analysis, similarity matrix
This article is included in the Cheminformatics gateway.
chemical space, data visualization, epigenetics, principal components analysis, similarity matrix
We discuss further in the Introduction, the differences of ChemMaps with other similar approaches.
We updated the Figures 1-3 for better visibility. Dataset 1 has been updated to also contain HDAC1 compounds used in the study.
We have expanded the perspectives of the work in the Conclusion.
The Supplementary File has been updated with Supplementary Methods, Supplementary Results and Table S1, containing the curation of the database and PCA details. Supplementary Figures S1-S4 have been revised, and we added a new Supplementary Figure 5 comparing the variance percentage contribution of the PCs for each studied database.
See the authors' detailed response to the review by Jean-Louis Reymond
See the authors' detailed response to the review by Dmitry I. Osolodkin
See the authors' detailed response to the review by Gerald Maggiora
Visual representation of chemical space has multiple implications in drug discovery for virtual screening, library design and comparison of compound collections, among others1. Amongst the multiple methods to explore chemical space, principal component analysis (PCA) of pairwise similarity matrices computed with structural fingerprints has been used to analyze compound datasets2,3. A drawback of this approach is that it becomes impractical for large libraries due to the large dimension of the similarity matrix4. Other approaches use molecular representations different from structural fingerprints, such as physicochemical properties or complexity descriptors, or methods different from PCA, such as multidimensional-scaling and neural networks5,6.
In representation of the chemical space based on PCA there have been “chemical satellite” approaches, such as ChemGPS, which select satellites molecules that might not be included in the database to visualize, but have extreme features that place them as outliers, with the intention to reach as much of the chemical space as possible7–10. Also, a related and more recent approach, Similarity Mapplet, makes possible the visualization of very large chemical libraries, by considering PCA of different molecular features, including structural11.
Although we concur with the fact that not all compounds in a compound data set should be necessary to generate a meaningful chemical space, there are still obvious limitations of using a fixed set of satellites to which the user is blinded. Also, until now there was no proposal of such a method based on structural similarity.
We therefore suggest the hybrid approach, ChemMaps, in which a portion of the database to be represented is used as satellite, thereby decreasing the computational effort required to compute the similarity matrix without losing adaptability of the method to any particular database. Since it is expected that more diverse sets would require more satellites, a second goal of this study was to qualitatively explore the relationship between the internal diversity of compound datasets and the fraction of compounds required as satellites, in order to generate a good approximation of the chemical space.
Table 1 summarizes the six compound data sets considered in this study. Note that small median similarity values imply higher diversity. The datasets were selected from a large scale study of profiling epigenetic datasets (unpublished study, Naveja JJ and Medina-Franco JL) with relevance in epigenetic-drug discovery. We also included DrugBank as a control diverse dataset12. Briefly, we selected focused libraries of inhibitors of DNMT1 (a DNA-methyltransferase; library diverse 2D and 3D), L3MBTL3 (a histone methylation reader; diverse 3D and less diverse 2D), SMARCA2 (a chromatin remodeller; diverse 2D, less diverse 3D), and CREBBP (a histone acetyltransferase; less diverse both 2D and 3D). Datasets were selected based on their different internal diversity (as measured with Tanimoto index/MACCS keys for 2D measurements and Tanimoto combo/OMEGA-ROCS for 3D; see Figure S1 in Supplementary File 1). Data sets in this work have approximately the same number of compounds except for HDAC1 and DrugBank, which were selected to benchmark the method in larger databases (Table 2). We evaluated 2D diversity using the median of Tanimoto/MACCS similarity measures in KNIME version 3.3.2, and 3D diversity using the median of Combo Score from the ROCS, version 3.2.2 and OMEGA, version 2.5.1, OpenEye software13–16.
| Dataset | Description | Size | 2D similaritya | 2D similarityb | 3D similarityc |
|---|---|---|---|---|---|
| DNMT1 inhibitors | DNA-methyltransferase | 244 | 0.44 | 0.12 | 0.16 |
| SMARCA2 inhibitors | Chromatin remodeller | 220 | 0.51 | 0.15 | 0.23 |
| CREBBP inhibitors | Histone acetyltransferase | 178 | 0.67 | 0.22 | 0.16 |
| L3MBTL3 inhibitors | Histone methylation reader | 115 | 0.77 | 0.41 | 0.03 |
| HDAC1 inhibitors | Histone acetyltransferase | 3,257 | 0.49 | 0.16 | 0.12 |
| DrugBank | Approved drugs | 1,900 | 0.35 | NC | NC |
To assess the hypothesis of this work we performed two main approaches A): Backwards approach: start with computing the full similarity matrix of each data set and remove compounds systematically; and B) Forward approach: start adding compounds to the similarity matrix until finding the reduced number of required compounds (called ‘satellites’) to reach a visualization of the chemical space that is very similar to computing the full similarity matrix. The second approach would be the usual and realistic approach from a user standpoint. Each method is further detailed in the next two subsections.
The following steps were implemented in an automated workflow in KNIME, version 3.3.217:
1. For each compound in the dataset with N compounds, generate the N X N similarity matrix using Tanimoto/extended connectivity fingerprints radius 4 (ECFP4) generated with CDK KNIME nodes.
2. Perform PCA of the similarity matrix generated in step 1 and selected the first 2 or 3 principal components (PCs).
3. Compute all pair-wise Euclidean distances based on the scores of the 2 or 3 PCs generated in step 2. The set of distances are later used as reference or ‘gold standard’. It should be noted that the “real” distances or true gold standard would consider the whole distance matrix. However, for visualization purposes it is unfeasible to render more than 3 dimensions. Therefore, we selected as reference the best 2D or 3D visualization possible by means of PCA.
4. Repeat steps 1 to 3 with one compound as satellite, generating an N X 1 similarity matrix. The first compound was selected randomly. In this case, for example, it is only possible to calculate one PC, but as the number of satellites increases, we can again compute 2 or 3 PCs.
5. Calculate the correlation among the pairwise distances generated in step 2 obtained using the whole matrix (e.g., gold standard) and those obtained in step 4.
6. Iterate over steps 4 and 5 increasing the number of satellites one by one until N - 1 satellites are reached. To select the second, third, etc. compounds, two approaches were followed: select compounds at random and select compounds with the largest diversity to the previously selected (i.e., Max-Min approach).
7. Estimate the proportion of satellite compounds required to preserve a ‘high’ (of at least 0.9) correlation.
8. The prior steps were repeated five times for each dataset in order to capture the stability of the method.
The former approach is useful only for validation purposes of the methodology as a proof-of-principle. However, the obvious objective of a satellite-approach is to avoid the calculation of the complete similarity matrix e.g., step 1 in backwards approach. To this end, we developed a satellite-adding or forward approach, in contrast with the formerly introduced backwards approach. We started with 25% of the database as satellites and for each iteration we added 5% until the correlation of the pairwise Euclidean distances remains high (at least 0.9). A further description of the methods for standardizing the chemical data and integrating the dataset can be found in the Supplementary material, as well as a further description of the PCA analysis used.
In this pilot study, we assessed a few variables to tune up the method, such as the number of PCs used (2 or 3) and the selection of satellites at random or by diversity. We found that selection at random is more stable, above all in less diverse datasets (Figure 1 and Figure 2; Figure S2 and Figure S3). Likewise, selecting 2 PCs the performance is slightly better and more stable (compare Figure 1 and Figure 2 against Figure S2 and Figure S3).
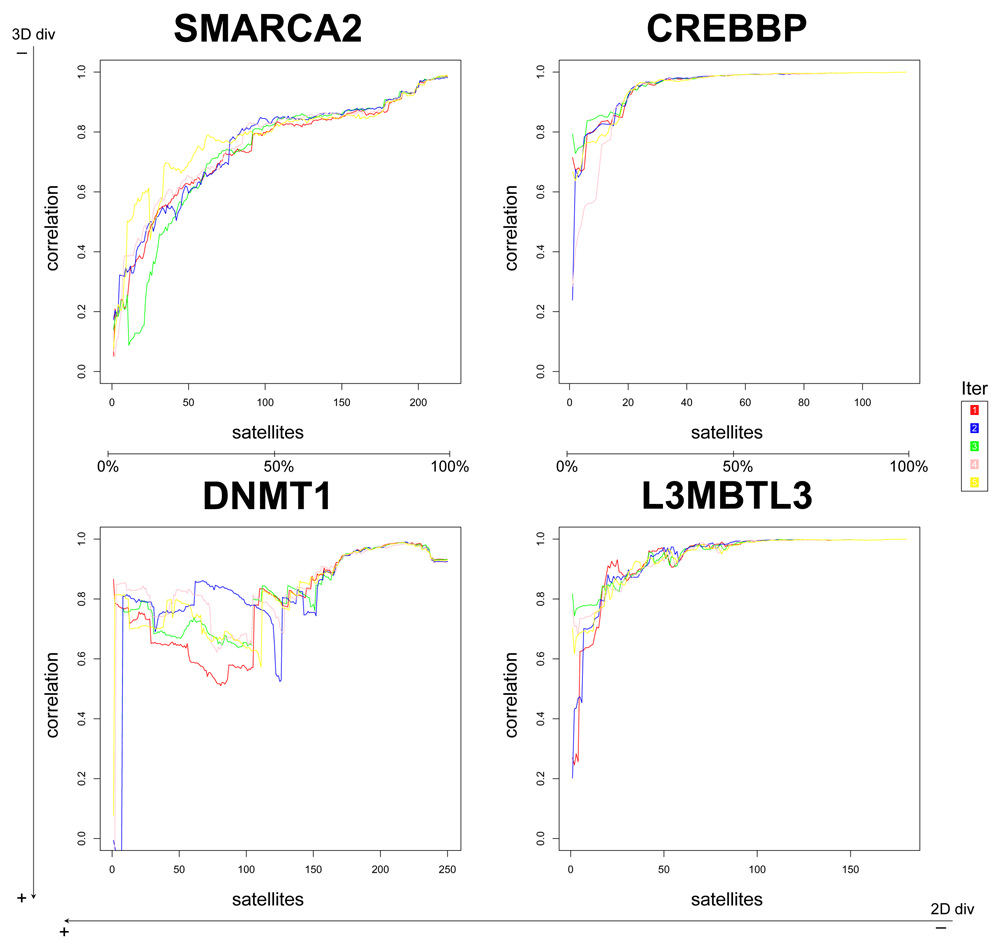
The correlation with the results from the whole matrix was calculated with increasing numbers of satellites. Each colored line represents one of the five iterations.
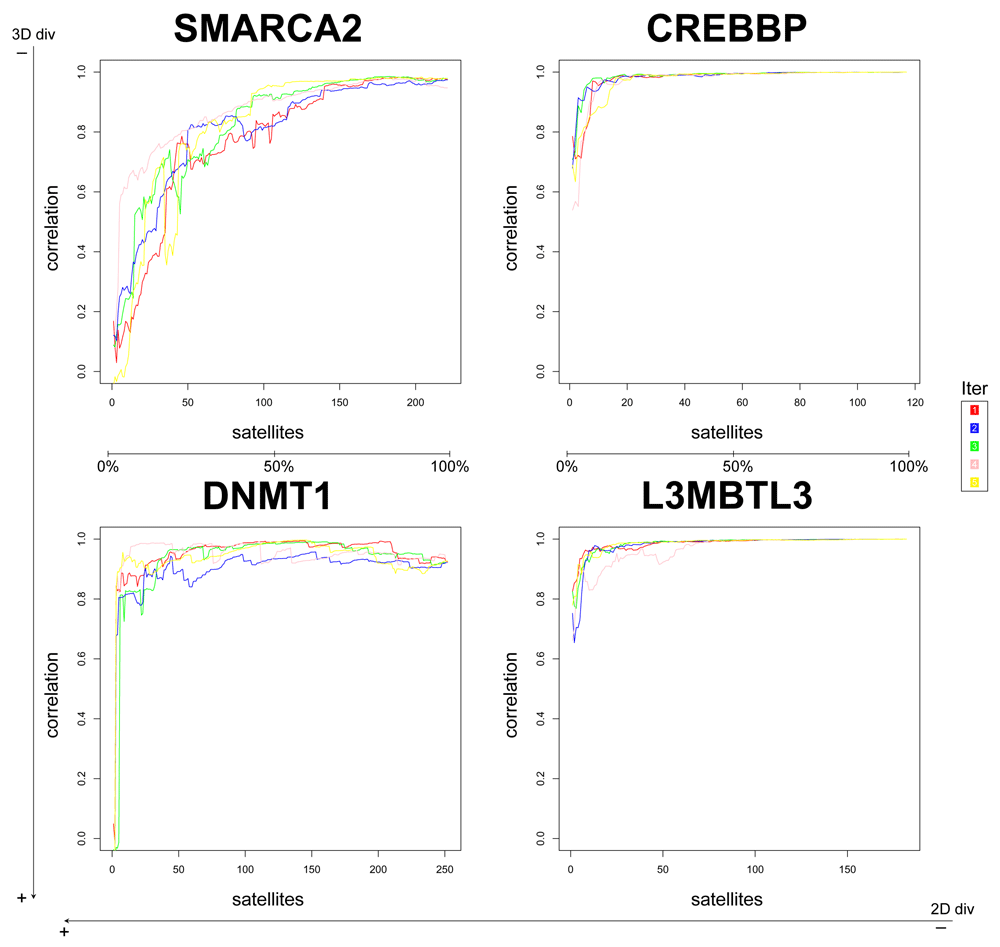
The correlation with the results from the whole matrix was calculated with increasing numbers of satellites. Each colored line represents one of the five iterations.
Therefore, from this point onwards we will focus on the results of the at random satellites selection and using 2 PCs (Figure 2). From the four datasets, we conclude that for datasets with lower 2D diversity (CREBBP and L3MBTL3, see Table 1), around 25% of satellite compounds are enough to obtain a high correlation (≥ 0.9) with the gold standard (e.g., PCA on the whole matrix), whereas for 2D-diverse datasets i.e., DNMT1 and SMARCA2, up to 75% of the compounds could be needed to ensure a high correlation. Nonetheless, even for these datasets, using 25% of the compounds as satellites the correlation with the gold standard is already between 0.6 and 0.8; using 50% of the compounds as satellites the correlation is between 0.7 and 0.9. Hence, the higher the diversity of a dataset (especially 2D), the higher the number of satellites required.
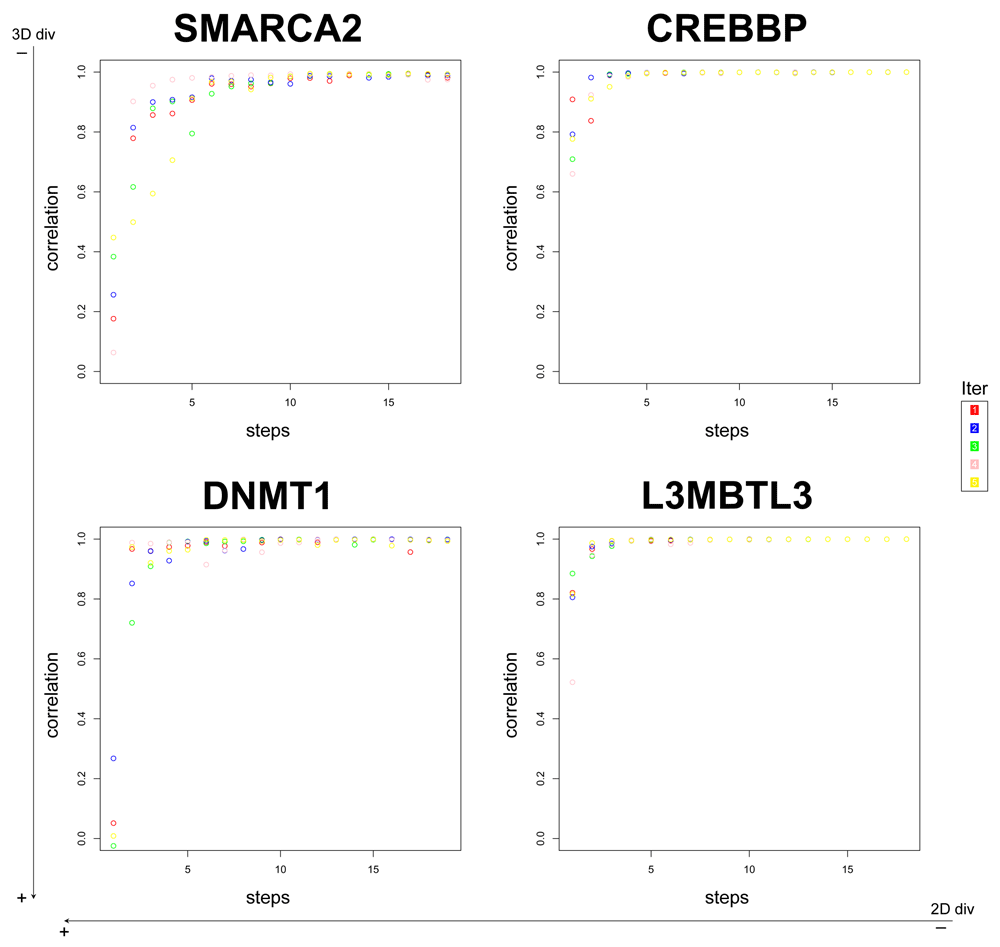
Evidently, a useful method for reducing computing time and disk space usage should not use the PCA on the whole similarity matrix to determine an adequate number of satellites for each dataset. With that in mind, we decided to design a method that starts with a given percentage of the database as satellites, and then keeps adding a proportion of them until the correlation between the former and the updated data is of at least 0.9. In Figure 3 we depict this approach on the same databases in Table 1 for step sizes of 5% and starting from zero. Similarly as what we saw in the backwards method, around 5 steps (25% of the database) are usually necessary to reach a stable, high correlation between steps. Figure S4 shows that for step sizes of 10% there is no further improvement. Therefore we suggest that the method should, for default, start with 25% of compounds as satellites and then keep adding 5% until a correlation between steps of at least 0.9 is reached.
In this pilot study we applied the ChemMaps method to visualize the chemical space of two larger datasets (HDAC1 and DrugBank with 3,257 and 1,900 compounds, respectively, Table 1). As shown in Table 2, a significant reduction in time performance was achieved as compared to the gold standard, and the correlation between the gold standard and the satellites approach was in both cases higher than 0.9. Figure 4 depicts the chemical spaces generated in both instances. Although the orientation of the map changed for HDAC1, the shape and distances remain quite similar, which is the main objective. This preliminary work supports the hypothesis that a reduced number of compounds is sufficient to generate a visual representation of the chemical space (based on PCA of the similarity matrix) that is quite similar to the chemical space of the PCA of the full similarity matrix.
| Database | Gold standard timing (s) | Satellites timing (s) | Correlation |
|---|---|---|---|
| DrugBank | 162 | 147 | 0.92 |
| HDAC1 | 406 | 287 | 0.99 |
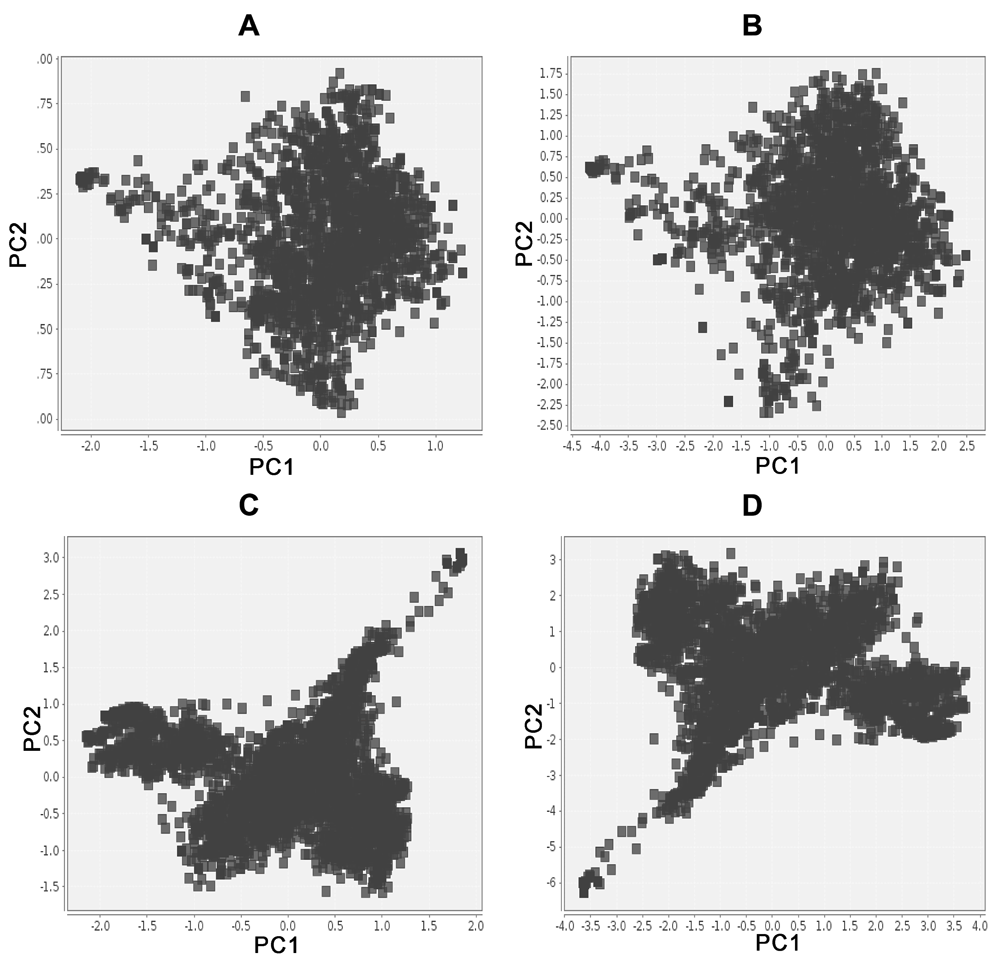
Chemical space of DrugBank using (A) the adaptive satellites approach or (B) the gold standard. As well as for HDAC1 using (C) the adaptive satellites approach or (D) the gold standard.
This proof-of-concept study suggests that using the adaptive satellite compounds ChemMaps is a plausible approach to generate a reliable visual representation of the chemical space based on PCA of similarity matrices. The approach works better for relatively less-diverse datasets, although it seems to remain robust when applied to more diverse datasets. For datasets with small diversity, fewer satellites seem to be enough to produce a representative visual representation of the chemical space. The higher relevance of 2D diversity over 3D in this study could be importantly related to the fact that the chemical space depiction is based on 2D fingerprints. Therefore, the performance of the methods depicting the chemical space based on 3D fingerprints could also be assessed.
A major next step is to conduct a full benchmark study to assess the general applicability of the approach proposed herein, and also in larger databases, in which we anticipate this method would be even more useful. A second step is to propose a metric that determines the number of compounds required as satellites for PCA representation of the chemical space based on similarity matrices. As well, it is pending the development of quantitative metrics for assessing the stability of the satellites selection and thus conclusively establish the superiority of at random satellite selection. Finally, a more comprehensive and in-depth study of this new methodology should be addressed, in order to further characterise its applicability domain, including a dataset diversity threshold above which the confiability of the approach decreases.
Dataset 1. This file contains the six compound datasets used in this work in SDF format. No special software is required to open the SDF files. Any commercial or free software capable of reading SDF files will open the data sets supplied. http://dx.doi.org/10.5256/f1000research.12095.d17163218
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Thank you for your comment. In the first version of the manuscript we cited three papers about ChemGPS published between 2005 and 2009 (references 7-9). In a ... Continue reading Dear Dr. Oprea:
Thank you for your comment. In the first version of the manuscript we cited three papers about ChemGPS published between 2005 and 2009 (references 7-9). In a revised version of our manuscript we will include the citation to the first paper you wrote about ChemGPS (J. Comb. Chem. 2001, 3, 157-166). Thanks also for your suggestion to compare directly ChemMaps with ChemGPS.
Thank you for your comment. In the first version of the manuscript we cited three papers about ChemGPS published between 2005 and 2009 (references 7-9). In a revised version of our manuscript we will include the citation to the first paper you wrote about ChemGPS (J. Comb. Chem. 2001, 3, 157-166). Thanks also for your suggestion to compare directly ChemMaps with ChemGPS.