Keywords
Cav1.2, calpain cleavage, neuronal calcium
This article is included in the Preclinical Reproducibility and Robustness gateway.
Cav1.2, calpain cleavage, neuronal calcium
L-type Ca2+ channels are critical regulators of neuronal excitability (Berkefeld et al., 2006; Marrion & Tavalin, 1998), gene expression (Dolmetsch et al., 2001; Graef et al., 1999; Li et al., 2012; Ma et al., 2014; Marshall et al., 2011; Murphy et al., 2014; Wheeler et al., 2012), long-term potentiation (LTP) (Boric et al., 2008; Grover & Teyler, 1990; Moosmang et al., 2005; Patriarchi et al., 2016; Qian et al., 2017), long-term depression (LTD) (Bernard et al., 2014; Bolshakov & Siegelbaum, 1994), and memory consolidation (White et al., 2008). Cav1.2 is the most abundant L-type channel in the brain and heart (Hell et al., 1993a; Sinnegger-Brauns et al., 2004). The multitude of Cav1.2-dependent functions is illustrated by diseases such as Timothy Syndrome, which arises from one of three single missense mutations in exon 8/8A of the CACNA1C gene encoding the central, ion-conducting α11.2 subunit. Symptoms of this rare autosomal dominant disorder manifest as syndactyly, autistic-like behaviors, and widespread organ dysfunctions including dysregulation of cardiac contractility and heart rate (Splawski et al., 2004).
The central subunit of Cav1.2 that forms the ion-conducting pore, α11.2, exists in two major size forms with molecular masses estimated to be between 230-250 and 190-210 kDa (Bunemann et al., 1999; Davare et al., 1999; Davare & Hell, 2003; Davare et al., 2000; De Jongh et al., 1996; Hell et al., 1996; Hell et al., 1993a; Hell et al., 1995; Hell et al., 1993b; Kochlamazashvili et al., 2010; Patriarchi et al., 2016; Qian et al., 2017). CACNA1C was first cloned from rabbit heart, where full length α11.2 consists of 2171 residues with a predicted MR of 243 kDa (Mikami et al., 1989). Differential splicing of exons encoding the N-terminus of α11.2 and a number of other CACNA1C exons can result in isoforms that vary by 30 or more residues in length (Liao et al., 2015; Snutch et al., 1991, and references therein). Determination of the precise sizes of these α11.2 variants by SDS-PAGE is hampered by the fact that even a small increase in the concentration of acrylamide from 5 to 6 percent causes a strong change in migration of the two size forms (Hell et al., 1993b). These observations indicate that the migration behavior of α11.2 during SDS-PAGE can be anomalous.
Several studies over the past two decades detail the regulatory importance of calpain-mediated proteolysis at the α11.2 distal C-terminus (DCT) (Fuller et al., 2010; Hell et al., 1996; Hell et al., 1993b; Hulme et al., 2006b). For instance, deletion of 300-470 residues from the C terminus resulted in a 4-6 fold increase in current density without an increase in gating currents when expressed in Xenopus oocytes (Wei et al., 1994). These findings suggest that the potentiation due to C-terminal deletions is not caused by increased surface expression of Cav1.2, but by an increase in coupling of depolarization-induced movement of the voltage sensors to pore opening (Wei et al., 1994). Similarly, truncating α11.2 after residues 1733, 1821, 1905, and 2024 increased current density in HEK293-derived tsA201 cells by several fold, which was reversed by co-expression or injection of distal fragments as separate polypeptides (Fuller et al., 2010; Gao et al., 2001; Hulme et al., 2006b). Further deletions at or before residue 1623 abrogated channel currents, consistent with earlier work identifying residues 1623-1666 as critical for Cav1.2 surface expression (Gao et al., 2000). These latter findings are also in agreement with recent observations, in which binding of α-actinin to this region is important for Cav1.2 surface expression (Hall et al., 2013; Tseng et al., 2017).
Earlier evidence indicates that the 190-210 kDa short form results from proteolytic processing of the long form by the Ca2+-stimulated protease calpain (Hell et al., 1996). More recent work has suggested that extensive proteolytic processing occurs via calpain- and ubiquitin/proteasome-mediated mechanisms that target the intracellular loop between domains II and III, yielding two prominent α11.2 fragments: a 90 kDa fragment that might consist of the N-terminus and the first two integral membrane domains I and II, and a 150 kDa fragment that might consist of domains III and IV and the long C-terminus (Michailidis et al., 2014).
We performed a long overdue, systematic analysis of α11.2 size forms using region-specific antibodies, increasing concentrations of acrylamide, and surface biotinylation to examine their migration behavior during SDS-PAGE. As expected, one of the two main size forms of α11.2 migrates according to an apparent MR of 250 kDa, corresponding very well with the predicted size of the full length subunit. Importantly our study also provides very consistent and clear evidence that extensive proteolytic processing of α11.2 occurs within the last ~660 C-terminal residues, with minimal cleavage in the middle of the pore-forming portion of the channel. Although removal of the DCT would be expected to increase channel currents (Fuller et al., 2010; Wei et al., 1994), the severed DCT remains associated with the main channel portion to maintain a reduction of channel activity (Fuller et al., 2010; Gao et al., 2001; Hulme et al., 2006b).
We used 6–12 week old 50% C57black/6N and 50% 129Sv hybrid mice (Jackson Laboratories, Bar Harbor, MN), α11.2 conditional knockout (cKO) mice and their litter-matched WT controls as described (Patriarchi et al., 2016; White et al., 2008), and 8–12 week old Sprague Dawley rats (Harlan). CaV1.2 cKO mice of neuron-specific deletion and their wild-type littermates were on a C57BL/6NTac:129SvEv F2 genetic background. Mice with a floxed CaV1.2 exon two allele (CaV1.2 f/+ or CaV1.2 f/f) and maintained on a 129SvEv genetic background were first bred to transgenic mice expressing the Cre recombinase regulated by the synapsin 1 promoter (Syn1-CreCre/+) and maintained on a C57BL/6NTac background (Cui et al., 2008; Zhu et al., 2001), producing an F1 cross. Using non-littermate offspring from the F1 cross, heterozygous floxed, cre-positive (CaV1.2 f/+; Syn1-CreCre/+) mice were then crossed with heterozygous floxed, cre-negative (CaV1.2 f/+; Syn1-Cre+/+) mice to produce homozygous floxed, Cre-positive (CaV1.2 f/f; Syn1-CreCre/+) conditional knockout mice as well as wild-type mice (CaV1.2 +/+; Syn1-Cre+/+). All animals were housed by the Animal Care Unit in Tupper Hall at UC Davis. This facility is fully approved for NIH-funded research and accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. It maintains animals in a highly controlled environment optimized for the comfort of rodents in accordance with the applicable portions of the Animal Welfare Act and the DHS “Guide to the Care and Use of Animals.” Its NIH Office of Laboratory Animal Welfare Assurance Number is A3433-01. All efforts were made to ameliorate any potential suffering of animals. Specifically, animals were anesthetized with 5% isoflurane for 2–3 minutes in a two-chamber drop jar before decapitation and collection of tissue. This procedure followed NIH guidelines and was approved by the Institutional Animal Care and Use Committees at the University of California at Davis.
Residue numbers correspond to the initial α11.2 sequence from rabbit heart (Gene Bank Accession number: CAA33546).
The polyclonal antibody CNC1 was produced against the synthetic peptide (KY)TTKINMDDLQPSENEDKS, covering residues 818 to 835 within the intracellular loop between domains II and III of α11.2 (Dubel et al., 1992). The peptide was coupled to bovine serum albumin in the laboratory of W. A. Catterall (University of Washington, WA, USA) and used to immunize rabbits (Hell et al., 1993b). Before use, the antibody was affinity purified on the same peptide cross-linked to Sepharose 4B-CL (for validation and characterization of CNC1 see Davare et al., 1999; Hall et al., 2013; Hell et al., 1993a; Hell et al., 1993b). The lysine and tyrosine residues at the N-terminus had been added for cross-linking and labeling purposes.
The polyclonal antibody ACC-003 was obtained from the company Alomone Labs (catalog number ACC-003, batch number ACC003AN4725; Jerusalem, Israel). It was produced in rabbit against the synthetic peptide (C)TTKINMDDLQPSENEDKS, which like CNC1, covers residues 818 to 835 within the intracellular loop between domains II and III of α11.2. The cysteine at the N-terminus is not part of the original α11.2 sequence but had presumably been added for cross-linking purposes. The batch of this antibody we received was characterized in Figure 2.
The polyclonal antibody FP1 was produced against an N-terminal GST fusion protein covering residues 783 to 845 within the same intracellular loop between domains II and III of α11.2 as CNC1. The affinity purified GST fusion protein was used to immunize rabbits in the laboratory of J. W. Hell (University of Wisconsin, WI, USA). Before use, the antibody was affinity purified on the same GST fusion protein cross-linked to glutathione Sepharose (for validation and characterization see Davare et al., 2001; Davare et al., 2000; Hall et al., 2013; Hall et al., 2007; Hall et al., 2006).
The polyclonal antibody CNC2 antibody was produced against the synthetic peptide (KY)GRGQSEEALPDSRSYVS covering residues 2122-2138 of α11.2, a region ~40 residues upstream of the very C terminus of α11.2 (Hell et al., 1993b). The peptide was coupled to bovine serum albumin in the laboratory of W. A. Catterall (University of Washington, WA, USA) and used to immunize rabbits (Hell et al., 1993b). Before use, the antibody was affinity purified on the same peptide cross-linked to Sepharose 4B-CL (for validation and characterization see Davare et al., 1999; Hall et al., 2013; Hell et al., 1996; Hell et al., 1993b; Hulme et al., 2006a). The lysine and tyrosine residues at the N-terminus had been added for cross-linking and labeling purposes.
The phosphospecific polyclonal antibody against pS1700 was produced against the synthetic peptide EIRRAIpSGDLTAEEEL (residues 1694-1713) (Fuller et al., 2010). The peptide was coupled to bovine serum albumin in the laboratory of W. A. Catterall (University of Washington, WA, USA) and used to immunize rabbits. Before use, the antibody was affinity purified on the same peptide cross-linked to Sepharose 4B-CL (for validation and characterization see Fuller et al., 2010; Murphy et al., 2014).
The phosphospecific polyclonal antibody against pS1928 was produced against the synthetic peptide LGRRApSFHLECLK (residues 1923-1932) (Davare et al., 1999). The peptide was coupled to bovine serum albumin in the laboratory of W. A. Catterall (University of Washington, WA, USA) and used to immunize rabbits. Before use, the antibody was affinity purified on the same peptide cross-linked to Sepharose 4B-CL (for validation and characterization see Davare & Hell, 2003; Davare et al., 2000; Hall et al., 2007; Hall et al., 2006).
All procedures were performed on ice. Instruments, including centrifuge rotors, tubes, tools, and buffers, were pre-cooled at 4°C or on ice to minimize post-mortem proteolysis (Hell et al., 1993a; Hell et al., 1993b; Westenbroek et al., 1992). Whole mouse brains and acute rat forebrain and cortical slices were extracted with 1% Triton X-100 in 150 mM NaCl, 10 mM EDTA, 10 mM EGTA, 10 mM Tris, pH 7.4 containing protease inhibitors (0.1 mM phenylmethylsulfonyl fluoride, 1 µM pepstatin A, 2 µM leupeptin, 4 µM aprotinin) and phosphatase inhibitors (2 µM microcystin LR, 1 mM p-nitrophenyl phosphate, 1 mM sodium pyrophosphate, 2.5 mM sodium fluoride). Extracts were cleared by 30 minutes centrifugation (250,000xg). The soluble fraction was incubated on a head-over-head tilter with protein A - Sepharose beads and 2 µg FP1 antibody for 4 h at 4° C and washed three times with 0.1% Triton X-100 in 150 mM NaCl, 10 mM EDTA, 10 mM EGTA, 10 mM Tris, pH 7.4. Immunoprecipitated Cav1.2 underwent SDS-PAGE in gels with a stacking phase polymerized from 3.5% acrylamide and a separating phase polymerized from 5, 7, 9, 11, or 13% acrylamide. Protein was transferred to polyvinylidene fluoride (PVDF) membranes at 50 V for 600 minutes for subsequent probing as previously described (Davare et al., 1999; Hell et al., 1993a; Hell et al., 1993b). Briefly, membranes were blocked in 10% milk, incubated in affinity-purified primary antibody (FP1 1:800, CNC1 1:200, CNC2 1:50, anti-pS1700 1:400, anti-pS1928 1:100, ACC-003 1:400) for 2 hours, washed, incubated in horseradish peroxidase (HRP)-labeled Protein A for 1 hour, washed, and developed on autoradiography film using chemiluminescence.
Forebrain slices were prepared from rat brain, then non-cortical regions trimmed when indicated to obtain cortical slices, equilibrated in oxygenated (95% O2, 5% CO2) artificial cerebral spinal fluid (ACSF: 119 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl, 1 mM MgSO4, 2.2 mM CaCl2, 15 mM glucose, 1 mM myo-inositol, 2 mM Na-pyruvate, 0.4 mM ascorbic acid) at 32°C for 1 h, and labeled at 4°C for 45 min in 2 ml ACSF containing 1 mg/ml Sulfo-NHS-SS-biotin (Pierce). Oxygenation of all slices was maintained throughout the entirety of the experiment for slice equilibration, biotinylation, quenching and lysis procedures. Excess Sulfo-NHS-SS-biotin was quenched by washing slices four times with ice-cold ACSF buffer containing 100 mM glycine. Cells were homogenized on ice with 50 mM Tris-Cl pH 7.4, 150 mM NaCl, 10 mM EGTA, 10 mM EDTA, 1% NP-40, 10% Glycerol, 0.05% SDS, 0.4% DOC containing protease and phosphatase inhibitors and insoluble material removed by centrifugation (10,000 xg, 20 min). Biotinylated constituents in lysate, each containing 300 μg of protein, were affinity-purified by incubation with 30 µl of NeutrAvidin-conjugated Sepharose beads (Thermo-Fisher) for 3 h at 4°C. Following four ice-cold washes of bead-bound material with 1% Triton X-100, 150 mM NaCl, 10 mM Tris-Cl, 10 mM EDTA, 10 mM EGTA, immobilized proteins were eluted by treatment with SDS sample buffer, separated by SDS-PAGE (8% resolving gel), and transferred to PVDF before immunoblotting as above.
To identify the main size variants of brain α11.2, we performed immunoblotting with three different antibodies made against the loop between domains II and III as well as three different antibodies raised against other various parts of the C-terminus of α11.2 (Davare et al., 2000; Hell et al., 1993a; Hell et al., 1993b) (Figure 1). FP1, CNC1, and the commercial antibody ACC-003 were raised against peptides covering middle portions of the II/III loop of α11.2. The anti-phospho-S1700 antibody (pS1700) was produced against the respective phosphopeptide covering residues 1694-1713 in the C-terminus, the anti-phospho-S1928 antibody (pS1928) against the respective phosphopeptide covering residues 1923-1932, and CNC2 against residues 2122-2138 near the very C-terminus of α11.2 (Figure 1).
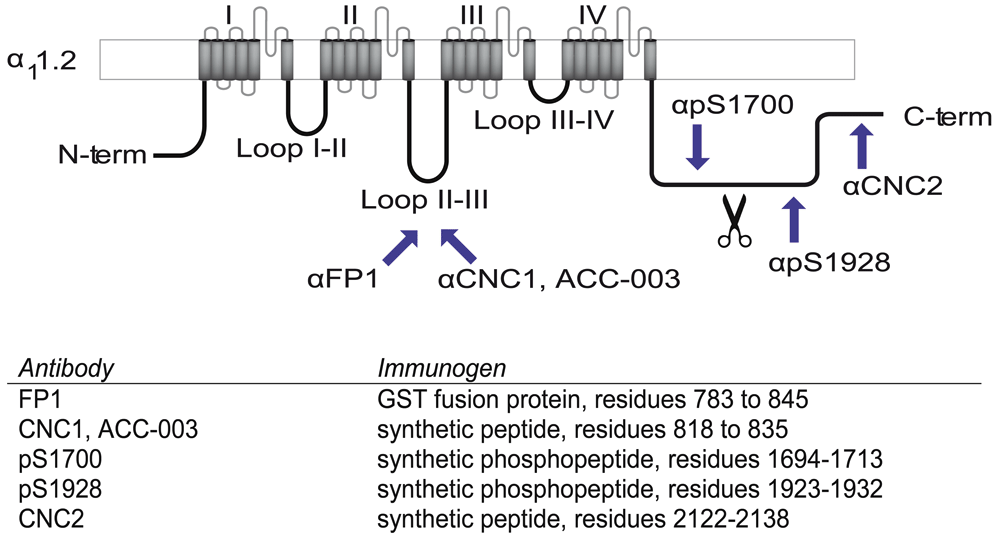
Shown is a schematic of the Cav1.2 α11.2 subunit, in which regions used as immunogens for the depicted antibodies are identified by arrows. Exact residues are listed in the table and numbered according to α11.2 given in Gene Bank Accession number CAA33546. FP1, CNC1, and ACC-003 are directed against the loop between domains II and III, pS1700 against phosphorylated S1700, pS1928 against phosphorylated S1928, and CNC2 against residues 2122-2138 of α11.2, which are ~40 residues upstream of the very C terminus of α11.2.
We tested whether immunoreactive bands recognized by these antibodies correspond to α11.2 size forms using brain extracts from WT and α11.2 KO mice. Total KO of α11.2 is embryonically lethal due to the central role of Cav1.2 triggering heart beat (Seisenberger et al., 2000). Thus we used tissue from conditional α11.2 KO mice (cKO) in which the floxed α11.2 gene was excised by Cre recombinase, whose expression was driven by the synapsin I promoter, resulting in a pan neuronal deletion throughout the brain (Cui et al., 2008; Zhu et al., 2001). We extracted whole mouse brain with 1% Triton X-100 (solubilizing >90% of total Cav1.2) and used the extracts directly for immunoblotting (Figure 2A). FP1 detected clear, strong bands of apparent MR of ~150, 210, and 250 kDa in WT mice. As expected for antibodies with immunoreactivity to α11.2, these 210 and 250 kDa bands were not readily detectable when cKO brain tissue was probed with FP1. Accordingly, these bands constitute bona fide α11.2 size forms. In contrast, the 150 kDa band was not only prominent in WT samples but also highly expressed in cKO brain, suggesting that this band does not correspond to α11.2 sequences. This conclusion is further supported when similar blots were probed with CNC1, which only recognized bands of 210 and 250 kDa in WT brain, both of which were undetectable in immunoblot lanes containing lysate from cKO mice. The ACC-003 antibody, a commercial antibody designed against the same epitope, recognized similar 210 and 250 KDa bands present in WT but not cKO brains, which is again consistent with these bands representing true major α11.2 size forms. However, this antibody detected additional immunoreactive bands of ~130 and ~190 kDa that were of equal strength in brain lysates from both WT and cKO mice, indicating that these two bands are not true isoforms of α11.2.
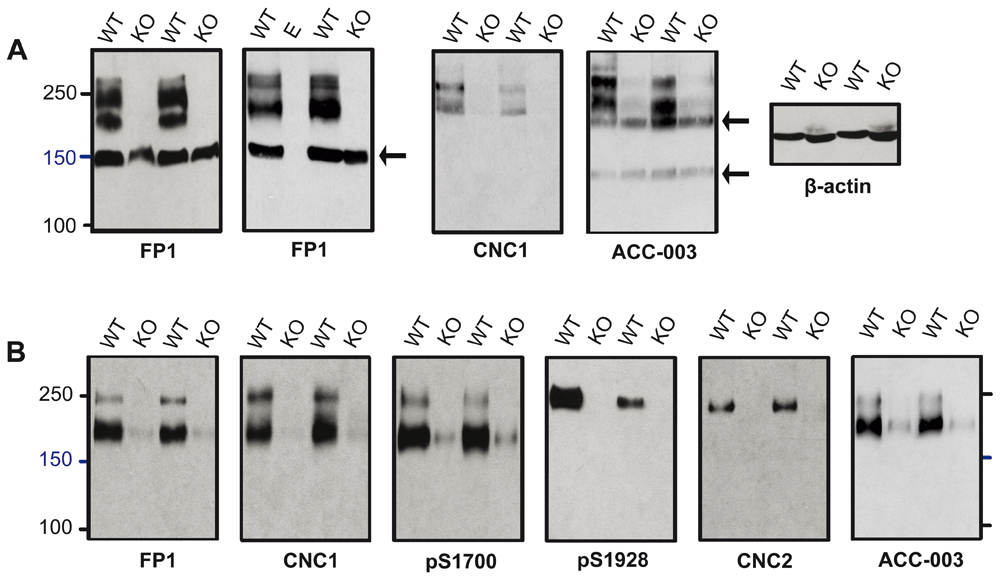
(A) Immunoblots of Triton X-100 extracts from conditional α11.2 KO mice (KO) and litter matched WT mice using gels polymerized from 8% acrylamide. To ensure that there was no spill-over between lanes, in some gels one or more lanes were left empty as shown here for the middle lane labeled E in the right FP1 blot. To fully resolve α11.2 short and long forms, the 100 kDa marker was run close to the bottom except in the right panel. In this experiment, electrophoresis of the same extracts used for α11.2 immunoblotting was terminated before the dye front reached the bottom. Probing for β-actin showed that comparable amounts of protein were present in each extract from the different WT and KI mice. (B) Cav1.2 was immunoprecipitated from brain extracts from conditional KO and WT mice with the FP1 antibody before SDS-PAGE in gels polymerized from 6% acrylamide and immunoblotting with the indicated antibodies. To fully separate α11.2 short and long forms, electrophoresis was performed until the 100 kDa marker was near the bottoms of the gels. For all antibodies, the ~210 and 250 kDa bands were nearly or completely absent in cKO samples.
For increased sensitivity and to further define the identity of the 150 kDa band detected in FP1 blots and the 130 and 190 kDa bands recognized by ACC-003, we performed immunoprecipitation to concentrate the α11.2 isoforms from a much larger volume of lysate. The FP1 antibody (of which we have a significantly larger supply than of the other antibodies) was used to immunoprecipitate α11.2 from Triton X-100 brain extracts. The resulting concentrate was then subjected to individual immunoblot analysis using the six distinct α11.2 antibodies available. Remarkably, probing with FP1 only revealed a 210 and a 250 kDa band but not the 150 kDa band (Figure 2B). Apparently this 150 kDa band detected by FP1 immunoblot of directly loaded brain extracts is not readily immunoprecipitated by FP1. This observation further suggests that the 210 and 250 kDa bands are immunologically different from the 150 kDa band, with the 210 and 250 kDa proteins but not the 150 kDa protein being efficiently immunoprecipitated. Moreover as with FP1, the CNC1, ACC-003, and pS1700 antibodies all recognized bands of 210 and 250 kDa in FP1 WT brain immunoprecipitates, whereas the more C-terminal directed pS1928 and CNC2 antibodies recognized only a single band of 250 kDa (Figure 2B). FP1, CNC1, ACC-003, and pS1700 immunoblotting did, as expected, reveal faintly reactive 210 and 250 kDa bands after FP1 immunoprecipitation from cKO brains. These weakly immunoreactive bands are the result of the continued α11.2 expression in non-neuronal tissue (glia and vasculature). Importantly, the 130 and 190 kDa bands recognized by ACC-003 in brain lysate of WT and cKO mice were not detectable after the FP1 immunoprecipitation. Similar to our observation that the 150 kDa band detected by FP1 probings of directly loaded brain lysates is not detected in blots of FP1 immunoprecipitates, this finding further indicates that the 130 and 190 kDa bands are not α11.2 isoforms.
Not all proteins, including MR markers, consistently migrate at the same apparent molecular mass during SDS-PAGE. It is conceivable that a protein of a true MR of 150 kDa could run with an apparent MR of 200 kDa and more. To increase certainty about the MR of the apparent 210 and 250 kDa bands detected in the above experiments and scrutinize whether the apparent 210 kDa band might under different conditions migrate near a 150 kDa marker, α11.2 migration relative to two different MR marker sets was analyzed in gels made from different concentrations of acrylamide (5–11%). For this analysis, Cav1.2 was enriched by immunoprecipitation with FP1. The individual marker proteins in the two different MR marker sets migrated uniformly and as expected for their molecular mass. Here, all five of the tested α11.2 antibodies recognized a protein band that migrated with the 250 kDa size markers in 5% gels and slightly slower than the 250 kDa markers in all other % acrylamide gels (Figure 3). The two loop antibodies FP1 and CNC1, as well as pS1700, but not pS1928 nor CNC2, recognized a second band that migrated either between the 150 and 250 kDa markers in 5% acrylamide gels or just below the 250 kDa markers in 7% gels, or co-migrated with the larger size form in 9, 11, and 13% gels. The pS1928 and CNC2 antibodies only detected the long form in brain extracts while the pS1700 antibody recognized both size forms, a pattern indicating that the shorter form represents an α11.2 size variant that is truncated, relative to full length, between residues 1700 and 1928. This notion is consistent with a size difference between the long and short forms of roughly 30–60 kDa and is also in agreement with the observed migration for the lower FP1-, CNC1-, and pS1700 immunoreactive band in 5–7% gels (the phospho-serine 1700 being 471 residues upstream of the distal C-terminus of full length α11.2).

Cav1.2 was immunoprecipitated from mouse brain extracts (Triton X-100) with the FP1 antibody against α11.2 before fractionation by SDS-PAGE in gels polymerized from 5, 7, 9, 11, and 13% acrylamide followed by immunoblotting with the indicated antibodies. Two different prestained marker protein sets were used to estimate MR.
In some cases, a faint immunoreactive band with an apparent MR of ~130 kDa in 5% gels and ~150 kDa in 7%, 11% and 13% gels was observed by immunoblotting with CNC1 and FP1. Figure 3 shows the clearest examples among all our immunoblots for detection of this weak band by CNC1 and FP1. However, in the majority of experiments a similar sized band was not detectable.
Given the anomalous migration of the short form, we wanted to provide further evidence for the estimation of a 30-60 kDa difference between the two size forms. α11.2 was expressed in HEK293 cells as either its full length form or as a shortened version truncated at residues 1800 (α11.2Δ1800) before extraction, immunoprecipitation and separation by 7% SDS-PAGE. As with the mouse brain lysate samples, FP1 and pS1700 detected full length and truncated α11.2 with an apparent MR of about 250 and 210 kDa, respectively, whereas the pS1928 antibody only identified the full length α11.2 (Figure 4A).
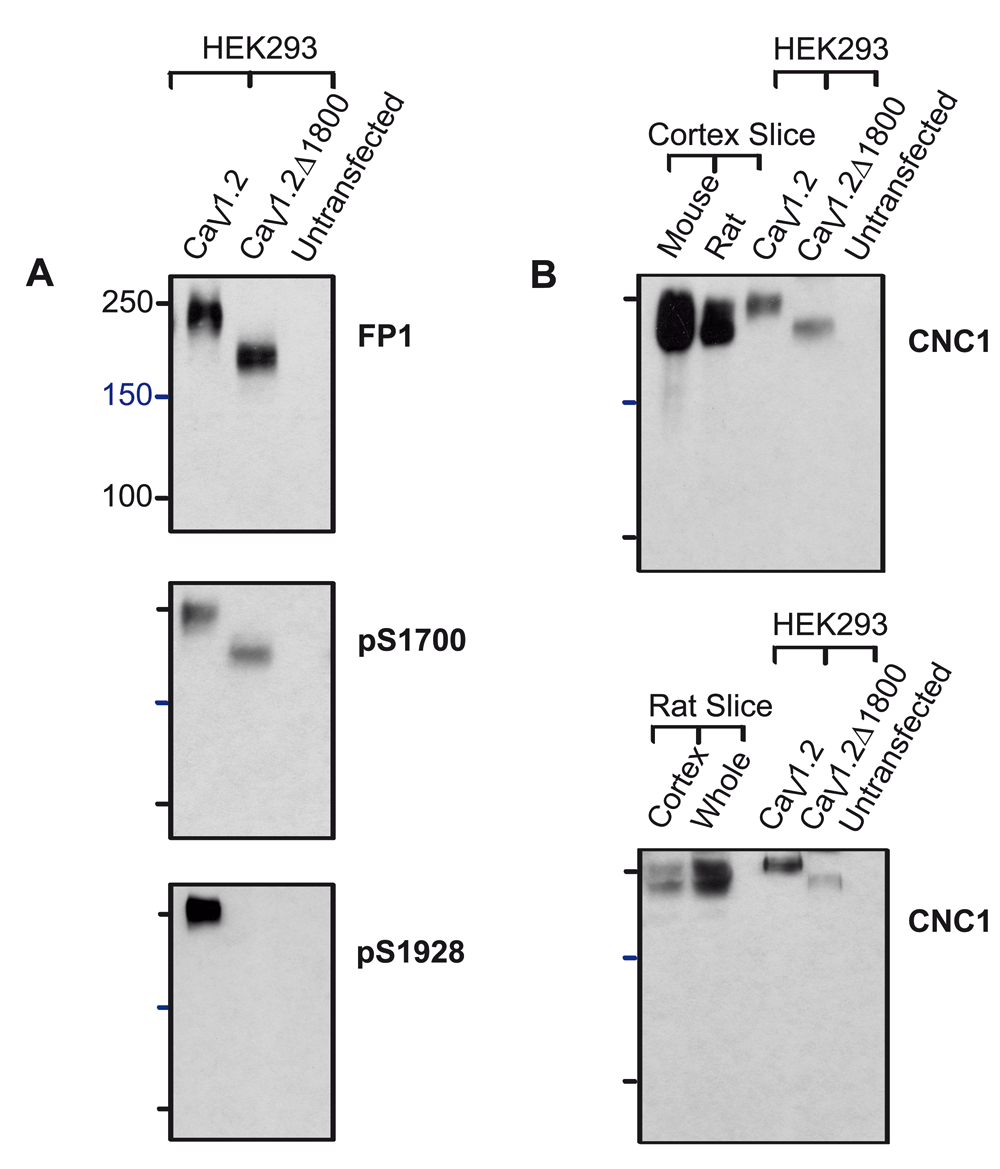
HEK293T cells were transfected with full length or truncated (Δ1800) α11.2 plus α2δ1 and β2a. HEK293T cells and rat and mouse brain slices were extracted with 1% Triton X-100 before immunoprecipitation of α11.2, SDS-PAGE in gels polymerized from 8% acrylamide, and immunoblotting with the indicated antibodies. (A) The full length form of α11.2 expressed in HEK293 cells migrated with an apparent MR of 250 kDa and is detected by FP1, pS1700 and pS1928. Truncated Δ1800 α11.2 migrated with an apparent MR of 210 kDa and is detected by FP1 and pS1700 but not pS1928. (B) The α11.2 short and long form appear only partially resolved because the weak α11.2 signals in HEK293 cell samples required long exposure times. The upper band as detected by CNC1 after FP1 immunoprecipitation from rat and mouse forebrain slices and cortical slices co-migrated with the full length form of α11.2 expressed in HEK293 cells, while the lower band co-migrated with the truncated Δ1800 α11.2 expressed in HEK293 cells.
Additional experiments were performed with rat tissue to look for potential differences in proteolytic processing between mouse and rat α11.2. We extracted forebrain slices and cortical slices from both mouse and rat for immunoprecipitation with FP1 and separation by SDS-PAGE, matching the 8% acrylamide gel conditions used in (Michailidis et al., 2014). As expected from our earlier analysis in 7 and 9% acrylamide gels, the α11.2 short form was partially separated from the long form in the 8% acrylamide gel (Figure 4B). Importantly, the long and short forms from the rodent brain tissues co-migrated with the corresponding full length α11.2 and α11.2Δ1800 ectopically expressed in HEK293 cells. Accordingly, truncation of the long form at approximately residue 1800 is most likely what gives rise to the main α11.2 short form in rodent brain. Moreover, these experiments did not reveal a protein band isolated from rat brain lysates that could conceivably correspond to a 150 kDa size form of α11.2, and only a very weak band of ~150 kDa could be detected in the mouse samples.
To test whether pull-down of surface biotinylated proteins might enrich for a unique α11.2 population at the cell surface and thereby unmask a size form smaller than 200 kDa, we performed surface biotinylation of acute slices using acute slices made from both total rat brain and cortex before extraction. We then carried out neutravidin-Sepharose pulldown and immunoblotting as described earlier (Michailidis et al., 2014). In agreement with our findings above, CNC1 and FP1 immunoblotting of proteins in neutravidin-Sepharose pulldowns and total lysate loads separated by 8% PAGE revealed major partially separated bands at ~200–250 kDa and no evidence of a 150 kDa band (Figure 5). On some immunoblots a weak band within the 90 kDa range was detectable by CNC1 (Figure 5B). Neutravidin pulldowns of unbiotinylated control samples did not yield detectable immunoblot signals, verifying the specificity of the biotin-neutravidin pulldown assay.
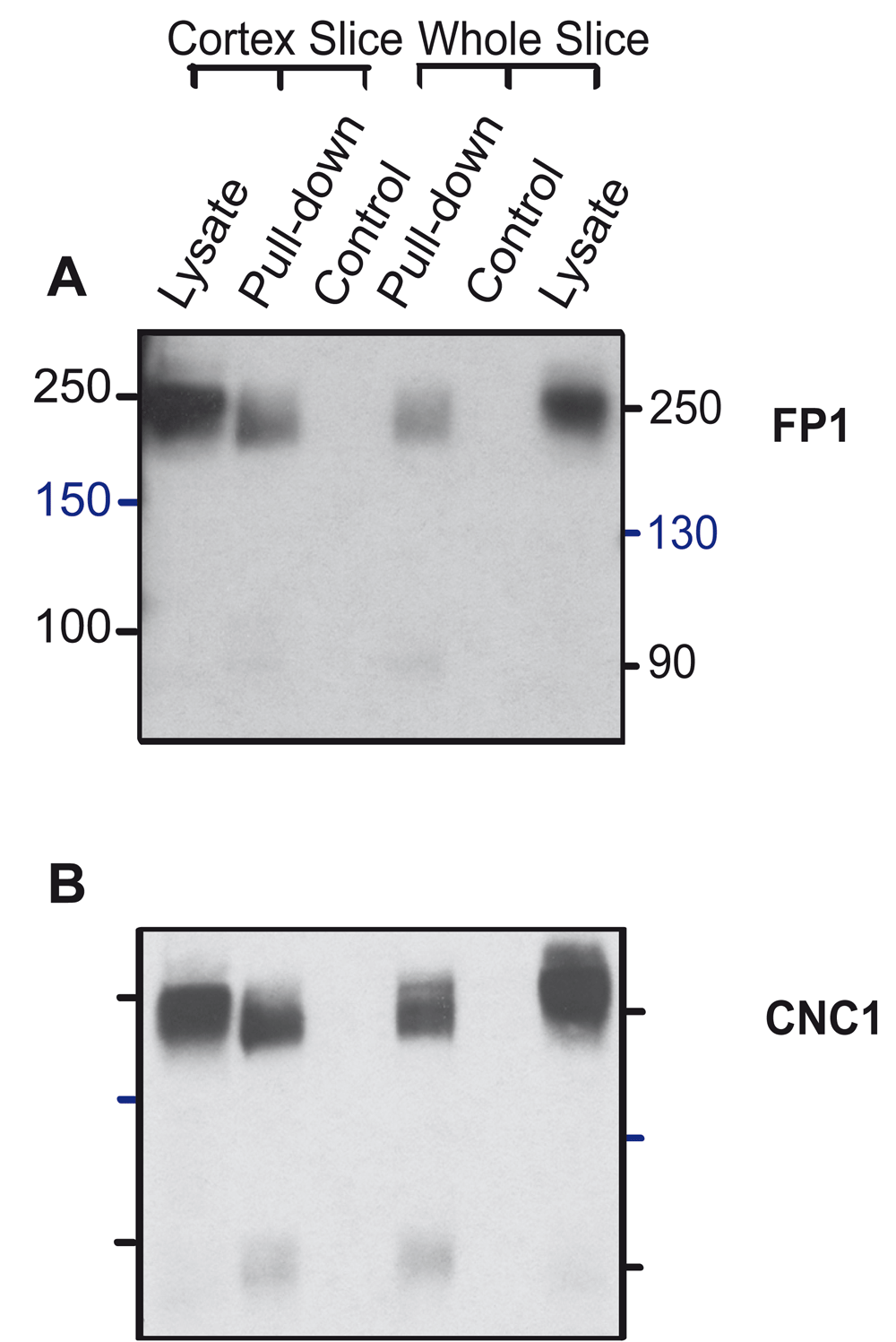
Cortical and forebrain slices were surface biotinylated and solubilized before pulldown with NeutrAvidin Sepharose, SDS-PAGE in 8% acrylamide gels, and immunoblotting with CNC1 and FP1. Control reflects slices mock treated without Sulfo-NHS-SS-biotin to demonstrate specificity of pulldown. Twenty μL lysate was also directly loaded for comparison.
Because we observed in some experiments a weak ~150 kDa band in FP1 immunoprecipitates that were immunoblotted with FP1 and CNC1 (Figure 3), we wanted to clarify whether this band is related to the strong 150 kDa band detected with FP1 in brain lysate of WT and cKO mice. We ran in parallel forebrain extracts and FP1 immunoprecipitates on the same gel (Figure 6). As before (Figure 2), CNC1 did not detect a 150 kDa band in lysate lanes (Figure 6A) even when blots were exposed to film for longer time periods (Figure 6B). However upon prolonged film exposure CNC1 probed blots reveal a faint 150 kDa band in lanes for the FP1 immunoprecipitated samples isolated from WT mice. Extended film exposure also revealed a weak 150 kDa band detected by FP1 after immunoprecipitation with FP1 (Figure 6C). Because the faint band in FP1 immunoprecipitates is equally well detected by FP1 and CNC1 but the strong 150 kDa band seen with FP1 in lysate is only detected by FP1, the two ~ 150kDa bands are most likely not related to one another but rather represent different protein species. If these ~150kDa bands were the same protein the CNC1 antibody should detect the strong 150 kDa band in lysate as well. Finally, only a faint 150 kDa band was also detected by the ACC-003 antibody probe upon extended exposure of the blot to film (Figure 6E).
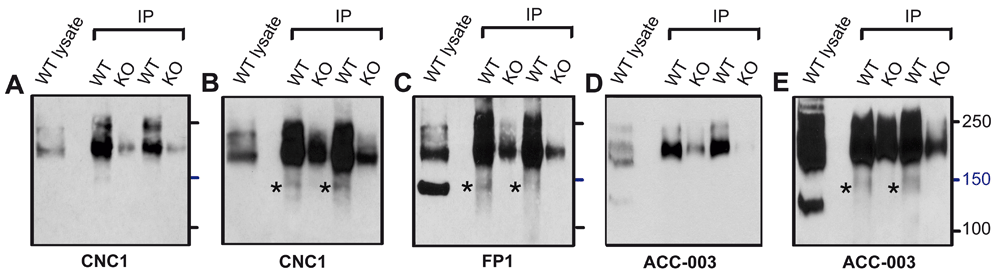
Immunoblots with CNC1 (A,B), FP1 (C), and ACC-003 (D,E) of Triton X-100 extracts from WT mice (lysate) and after immunoprecipitation with FP1 from cKO and WT mice. Gels were polymerized from 8% acrylamide. Note that a weak 150 kDa band is detected by CNC1, FP1, and ACC-003 after enrichment of α11.2 by immunoprecipitation with FP1 but the strongly immunoreactive 150 kDa band detected by FP1 in lysate is not detectable by either CNC1 or ACC-003.
Our extensive and detailed biochemical analysis of α11.2 size forms was inspired by recent work that suggested that surface α11.2 is cleaved to a large degree between domains II and III (Michailidis et al., 2014). The main evidence for the midchannel proteolysis proposed in this publication was based on immunoblotting:
1. The anti-LII-III antibody (ACC-003 from Alomone), detected two main bands that migrated with apparent MR values of ~150 and ~250 kDa (like CNC1, ACC-003 was made against α11.2 residues 818-835 in the loop between domains II and III);
2. An antibody produced against residues 2127-2143 near the very C-terminus of α11.2 (anti-LCt) recognized ~9 bands of varying intensities, one of which exhibited intermediate labeling intensity at an apparent MR of ~150 kDa;
3. An antibody against the N-terminus of α11.2 (anti-LNt) detected ~8 bands of varying intensities, including one of strong intensity that migrated with an apparent MR of ~90 kDa.
These observations are consistent with the possibility that cleavage could occur just N-terminal to the recognition site of ACC-003 / anti-LII-III inside the loop between domains II and III (Michailidis et al., 2014). The 250 kDa fragment recognized by ACC-003 / anti-LII-III would reflect the full length channel and the 150 kDa fragment recognized by ACC-003 / anti-LII-III would represent a fragment that comprises most of loop II/III, domains III and IV, and the full length C-terminus. The 90 kDa band detected with the N-terminal antibody would be the other cleavage product of the proposed midchannel cleavage and the anti-LCtrecognized 150 kDa band would be the remaining C-terminal cleavage product. However, it remains untested and unclear whether the N- and C-terminal antibodies in this work did indeed recognize their intended target and which among the many bands detected by these antibodies were truly α11.2, and not cross reactive proteins. Moreover, the 150 kDa band recognized by the anti-LCt antibody was a minor fraction of all the many bands detected by the anti-LCt antibody whereas the 150 kDa band recognized by the ACC-003 / anti-LII-III antibody was one of two major bands detected by the anti-LII-III antibody, making it unlikely that those two 150 kDa bands originated from the same protein.
One potential explanation for the detection of an apparent 150 kDa form of α11.2 (Michailidis et al., 2014) is that the full length α11.2 form and the C-terminally truncated form that we identify as 210 kDa in size were well separated (as in our 5% gels) but the 150 kDa MR marker ran slower in their experiments than expected, which is possible for pre-stained markers. It is also possible that the 210 kDa form ran faster than anticipated or that a combination of both occurred. These effects would result in an apparent MR value of our 210 kDa band that is less than the actual MR. Consistent with this possibility, the N-terminal antibody used in the previous work (Michailidis et al., 2014) recognized in addition to the 90 kDa band a 150 kDa band, which could be an overly fast migrating 210 kDa polypeptide. Importantly, by demonstrating precise co-migration of the short form with α11.2Δ1800 ectopically expressed in HEK293 cells, we ruled out the possibility that the short α11.2 form we identified with an apparent MR of ~210 kDa is actually a significantly smaller fragment (potentially with an MR of 150 kDa) that ran slower than would be expected for a polypeptide with an MR substantially below 210 kDa (Figure 4). Thus, the short α11.2 form we observed following isolation from rodent brains lacks ~371 C-terminal residues of full length α11.2, as is the case for α11.2Δ1800.
Based on our analysis of cKO brain extracts, the most likely explanation is that the earlier 150 kDa band detected by Michailidis et al. (Michailidis et al., 2014) was not a significant α11.2 isoform but rather a different protein recognized by the ACC-003 / anti-LII-III loop antibody. In fact, in addition to the 210 and 250 kDa bands seen only in α11.2 WT tissue and thereby reflecting major α11.2 size forms, the ACC-003 antibody we obtained from Alomone Labs did recognize an ~130 and an ~190 kDa band, which were present not only in α11.2 WT but also cKO mice. Similarly, another recent report indicates that the ACC-003 used in that work detected a band of ~130 kDa that was equally present in α11.2 WT and cKO tissue when the 250 kDa band was only present in WT but not cKO tissue (Bavley et al., 2017). It is unclear whether the 150 kDa band recognized by the ACC-003 / anti-LII-III antibody in the earlier work (Michailidis et al., 2014) corresponds to the 130 kDa band we detect with the ACC-003 antibody. This explanation is quite conceivable as migration behavior of native proteins (and even MR markers) can easily vary between gel systems, as we showed for the ~210 kDa α11.2 size form in Figure 2 and discussed in the preceding paragraph. Alternatively, cross-reactivity of antibodies with proteins other than α11.2 could be different for the ACC-003 / anti-LII-III antibody batch used more than 2 years ago (Michailidis et al., 2014) and the ACC-003 antibody we received in 2016 from Alomone Labs. Such differences could be due to different immune system responses within the individual rabbits used for immunization at different times. This possibility would also explain why the ACC-003 antibody we obtained from Alomone Labs recognized a cross-reacting 190 kDa band when the earlier ACC-003 / anti-LII-III antibody did not (Michailidis et al., 2014).
In further support of the notion that antibodies against peptides derived from the LII-III loop of α11.2 can cross react with other proteins, our FP1 antibody recognizes a 150 kDa band of equal strength in extracts from WT and cKO brains, whereas the 210 and 250 kDa bands are strong in WT extracts and very faint in cKO extracts, the latter reflecting α11.2 expression in non-neuronal tissue and cells (Figure 2A). FP1 was made against a polypeptide spanning residues 783-845, which includes all of the residues of the synthetic peptide used to make the ACC-003 / anti-LII-III antibody, as well as our CNC1 antibody (residues 821-838). Perhaps, the 821-838 segment mimics not only the α11.2 epitope but also to some degree, though not perfectly, a related epitope on another protein that is present in WT and cKO mice. In fact, another antiserum that was produced completely independent from our CNC1 antibody but utilized the very same α11.2 peptide sequence also detected an ~150 kDa band of similar intensity in brain lysates from WT and cKO mice (Tippens et al., 2008). Of note, the cKO mice used by Tippens et al. are different from the cKO used by us indicating that the strong 150 kDa band is present in mice of several different genetic backgrounds. Concordant with this idea, neither the ACC-003 antibody that we received from Alomone Labs that recognized a 130 kDa band in WT and cKO brains nor our CNC1 antibody recognized the strong 150 kDa band seen with FP1 in brain lysate. This finding is, once more, likely due to variability in immune responses of the individual rabbits to the immunogen, which at times but not always gives rise to antibodies against this unknown 150 kDa protein.
The results of our rigorous testing and validation of the antibodies used herein (see also Hall et al., 2013) boosts our confidence that the ~250 kDa protein detected by all six antibodies and the ~210 kDa protein detected by the four antibodies that recognize epitopes upstream of residue 1800 are two different size forms of α11.2. In contrast, the ~130 and ~150 kDa bands detected with ACC-003 / anti-LII-III and FP1, respectively, are most likely not related to α11.2 as these bands persist in α11.2 cKO tissue. Overall, the evidence is overwhelming that the prominent bands in the 130-150 kDa range detected by the various anti-α11.2 antibodies represent proteins that are different from α11.2.
Theoretically, it is also possible that the 150 kDa protein species arose because of nonspecific post mortem proteolysis. Because we use a strong and well-defined cocktail of inhibitors against serine, cysteine, and metalloproteases (Hell et al., 1993a; Hell et al., 1993b; Westenbroek et al., 1992) (see Material and Methods) and are particularly careful to keep all samples cold, such proteolysis may not have occurred to a significant degree in our hands. Accordingly, we only detected at best a very weak band migrating with an apparent MR of 150 kDa with the four different antibodies that recognized also full length α11.2. Under less stringent conditions, greater proteolysis might occur post mortem during tissue extraction, biotinylation, and purification. We tested whether incubation of forebrain slices at room temperature without O2 supply for 10 and 20 min would trigger proteolytic processing that results in a 150 kDa α11.2 band. However, in several different experiments we did not observe any increase in the weak 150 kDa band that is detected by either FP1 or CNC1 after enrichment of Cav1.2 by immunoprecipitation with FP1 (data not shown). Thus it appears unlikely that any 150 kDa band is due to post mortem proteolytic processing of α11.2.
If the 150 kDa band reported in the previous work (Michailidis et al., 2014) does not correspond to the 210 kDa fragment of α11.2 that arises via cleavage in the middle of the C-terminus, why then did Michailidis et al. not observe a doublet of 210 and 250 kDa in their hands with the ACC-003 / anti-LII-III antibody? Perhaps differences in SDS-PAGE procedures might result in the 250 and 210 kDa size forms not being separated at all during their analysis and instead appear to migrate as one band at 250 kDa, analogous to our finding that the two size forms co-migrate as a single band in 11 and 13% gels. This is possible even in 8% gels as the electrophoresis period applied by Michailidis et al. was most likely shorter than in our hands. For the analysis of α11.2 we reported here, the gel electrophoresis was extended to the point that the 60 kDa marker ran off the gel. Even with this protocol we see only partial separation of the 210 and 250 kDa forms of α11.2 in our 9% gels (Figure 4, Figure 5). With shorter running times, little to no separation is expected in 8% gels.
Immunocytochemical image analysis of ectopically expressed α11.2 that carries a GFP tag at its cytosolic N-terminus and an HA tag in one of the extracellular loops of domain III for anti-HA antibody labeling of surface expressed CaV1.2 was also used in the attempt to identify midchannel cleavage (Michailidis et al., 2014). The existence of clusters that only show GFP fluorescence is consistent with a substantial fraction of α11.2 being intracellular where HA labeling is absent. The existence of often very large HA-immunoreactive red clusters lacking GFP signals was interpreted as evidence for separation of GFP and HA tags by proteolysis. If so, channel halves would completely dissociate and not remain close to each other as would be required for a channel to function with modified current conductance. Accordingly, the potential for separate N- and C-terminal portions of α11.2 to form functional channels, as characterized by Michailidis et al. (2014), would either not be relevant in intact neurons if all of the cleaved channels dissociate or only apply to a small subpopulation of α11.2; however the degree and function of spatial separation of N- and C-terminal α11.2 fragments remains unclear.
Alternatively, rather than reflecting channel cleavage, the lack of detection of GFP signals in the HA-immunoreactive red clusters might be related to image acquisition or analysis. It is possible that the ectopically expressed α11.2, together with the GFP tag signal, is much higher inside dendritic shafts than at their surfaces, resulting in a steep gradient toward the periphery. If so, when images are taken so that the GFP signal in the center of the shaft is in the dynamic range (i.e., fairly strong but not saturated), the peripheral signal would be much weaker. This appears to be the case in Figure 2B and 2D in the preceding work (Michailidis et al., 2014), where GFP seems to be mostly in the center of the dendrite and HA, as expected for surface labeling, at the periphery while sparing the center. In this manner, GFP could appear weak or absent in peripheral areas of dendrites where HA is mostly localized due to the surface labeling for HA. Such a scenario would provide one potential explanation for surface areas showing strong HA and weak GFP signals where actually a sizable fraction of uncleaved α11.2 corresponding to the amount of HA signal might be present with the GFP signal at the surface appearing weak due to strong intracellular GFP signal.
Figure 2C in the Michailidis study (Michailidis et al., 2014) illustrates another potential scenario for dissociation of HA and GFP signals. This figure shows a long segment of the dendritic shaft that exhibits mostly HA and little if any GFP signal in. Even mild paraformaldehyde fixation can lead to permeabilization of 5–20 μm long segments of the dendritic plasma membrane and thereby expose sub-plasma membrane epitopes (Taylor & Fallon, 2006; Watschinger et al., 2008) (Matt and Hell, data not shown). Thus, it is conceivable that strong HA staining in this figure is paired with relatively strong suppression of GFP fluorescence in that segment as paraformaldehyde, which quenches GFP fluorescence, might have had preferential access to this region compared to the regions between the 0-2 and 12-14 μm marks where the GFP signal is much stronger. The strong HA staining in this dendritic segment could be surface labeling or intracellular HA staining of some sort of Cav1.2 clusters (perhaps reflecting a secretory compartment) due to antibody access induced by paraformaldehyde.
Evidence for the notion that HA staining likely yields much larger signals than GFP especially after fixation with paraformaldehyde is present in Figure 2D in this previous report (Michailidis et al., 2014). Here, protrusions are more strongly labeled by anti-HA staining than by GFP and shaft diameter appears much wider for HA than for GFP; these observations hint that the Cav1.2-GFP signal in or near the plasma membrane is rather weak and largely from intracellular Cav1.2. In this respect, it is surprising that there would be rather long segments of dendritic shaft that contain mostly HA and little if any GFP signal (as in Figure 2C of this publication).
We provide strong and clear evidence that the primary and major neuronal size forms of the α11.2 subunit of CaV1.2 are ~210 and 250 kDa in molecular mass. Based on detection of only a weak 150 kDa band by CNC1, ACC-003, and FP1 immunoblotting after immunoprecipitation with FP1 (Figure 3 and Figure 6), it appears that a very small fraction of α11.2 can be cleaved into 150 and 90 kDa fragments, which may remain to some degree associated with each other to form L-type channels of modified biophysical properties; however the prevalence of such proteolytic processing is certainly low (≤1%). It remains unclear what effects any limited mid-channel processing would have on overall L-type channel activity in neurons. It is possible that such processing of α11.2 is more prominent in certain cell types or subcellular regions and could in fact lead to the change in channel properties described by Michailidis et al. Determining where and under what condition(s) changes might occur will further rouse interesting questions for the future.
Dataset 1: Raw data supporting the findings presented in this study. The raw data shows full size film images of probed membranes. Full size membranes resulting from transfer of full size gels were often vertically cut to separate replicate sets of samples typically separated by MR markers for simultaneous probing of the different membrane fragments with different antibodies. For optimal resolution of the α11.2 long and short forms, which exhibit high MR, gels were run until the 60 kDa MR marker was either close to the very bottom of the gel or completely run off.
Raw data for Figure 2. Determination of antibody specificity for α11.2 with conditional α11.2 KO mice.
Original source images for Figure 2:
(A) Immunoblots of Triton X-100 extracts from conditional α11.2 KO mice (KO) and litter matched WT mice using gels polymerized from 8% acrylamide. To ensure that there was no spill-over between lanes, in some gels one or more lanes were left empty as shown here for the middle lane labeled E in the right FP1 blot. To fully resolve α11.2 short and long forms, the 100 kDa marker was run close to the bottom except in the right panel. In this experiment, electrophoresis of the same extracts used for α11.2 immunoblotting was terminated before the dye front reached the bottom. Probing for β-actin showed that comparable amounts of protein were present in each extract from the different WT and KI mice.
(B) Cav1.2 was immunoprecipitated from brain extracts from conditional KO and WT mice with the FP1 antibody before SDS-PAGE in gels polymerized from 6% acrylamide and immunoblotting with the indicated antibodies. To fully separate α11.2 short and long forms, electrophoresis was performed until the 100 kDa marker was near the bottoms of the gels. For all antibodies, the ~210 and 250 kDa bands were nearly or completely absent in cKO samples.
Raw data for Figure 3. Analysis of α11.2 size forms by SDS-PAGE with increasing acrylamide concentrations.
Original source images for Figure 3: Cav1.2 was immunoprecipitated from mouse brain extracts (Triton X-100) with the FP1 antibody against α11.2 before fractionation by SDS-PAGE in gels polymerized from 5, 7, 9, 11, and 13% acrylamide followed by immunoblotting with the indicated antibodies. Two different prestained marker protein sets were used to estimate MR.
Raw data for Figure 4. Mouse and rat α11.2 short forms co-migrate with α11.2 truncated at residue 1800 in the middle of the c-terminus.
Original source images for Figure 4: HEK293T cells were transfected with full length or truncated (Δ1800) α11.2 plus α2δ1 and β2a. HEK293T cells and rat and mouse brain slices were extracted with 1% Triton X-100 before immunoprecipitation of α11.2, SDS-PAGE in gels polymerized from 8% acrylamide, and immunoblotting with the indicated antibodies.
(A) The full length form of α11.2 expressed in HEK293 cells migrated with an apparent MR of 250 kDa and is detected by FP1, pS1700 and pS1928. Truncated Δ1800 α11.2 migrated with an apparent MR of 210 kDa and is detected by FP1 and pS1700 but not pS1928.
(B) The α11.2 short and long form appear only partially resolved because the weak α11.2 signals in HEK293 cell samples required long exposure times. The upper band as detected by CNC1 after FP1 immunoprecipitation from rat and mouse forebrain slices and cortical slices co-migrated with the full length form of α11.2 expressed in HEK293 cells, while the lower band co-migrated with the truncated Δ1800 α11.2 expressed in HEK293 cells. Sometimes, as seen here, a significant portion of the pore-forming subunit aggregated at the interface between stacking and resolving gels. This unresolved fraction (thick arrow) is not representative of its true molecular mass and not shown in the main figures.
Raw data for Figure 5. Surface biotinylation labels α11.2 size forms with apparent MR > 200 kDa in rat cortical and forebrain slices.
Original source images for Figure 5: Cortical and forebrain slices were surface biotinylated and solubilized before pulldown with NeutrAvidin Sepharose, SDS-PAGE in 8% acrylamide gels, and immunoblotting with CNC1 and FP1. Control reflects slices mock treated without Sulfo-NHS-SS-biotin to demonstrate specificity of pulldown. Twenty μL lysate was also directly loaded for comparison.
Raw data for Figure 6. Differential recognition of the strong 150 kDa FP1 band in lysate and weak 150 kDa band by FP1, CNC1, and ACC-003 after IP of α11.2 with FP1.
Original source images for Figure 6: Immunoblots with CNC1 (A,B), FP1 (C), and ACC-003 (D,E) of Triton X-100 extracts from WT mice (lysate) and after immunoprecipitation with FP1 from cKO and WT mice. Gels were polymerized from 8% acrylamide. Note that a weak 150 kDa band is detected by CNC1, FP1, and ACC-003 after enrichment of α11.2 by immunoprecipitation with FP1 but the strongly immunoreactive 150 kDa band detected by FP1 in lysate is not detectable by either CNC1 or ACC-003.
DOI, 10.5256/f1000research.11808.d168808 (Buonarati et al., 2017)
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)