Keywords
phosphatidylinositol-4,5-bisphosphate 3-kinase, p110, p85, insulin receptor substrate, insulin resistance, glucose intolerance
phosphatidylinositol-4,5-bisphosphate 3-kinase, p110, p85, insulin receptor substrate, insulin resistance, glucose intolerance
We have made the changes required by the referees for figure 1 and also added glucagon measurements to figure 4. Essentially, this means that more samples have been added and western blots quantified to make the results clearer and more convincing; datasets 1 and 4 have been updated.
See the authors' detailed response to the review by James R. Woodgett and Prital Patel
See the authors' detailed response to the review by Richard Z. Lin and Lisa Ballou
See the authors' detailed response to the review by Lily Q. Dong
Class IA phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) is a central mediator of a number of membrane receptor signaling pathways, including the insulin signaling pathway1,2. Following receptor activation by insulin, PI3K binds to tyrosine-phosphorylated amino acids of the insulin receptor substrates (IRS), resulting in PI3K activation and the formation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 has high affinity for the pleckstrin homology (PH) domain of the downstream target Akt/Protein Kinase B (PKB). The interaction of PIP3 with the PH domain enables phosphorylation of Akt/PKB by phosphoinositide dependent kinase (PDK) 1 and PDK 2, ultimately triggering a number of metabolic actions, such as lipogenesis, glycogen synthesis, inhibition of hepatic glucose output and increased glucose uptake in muscle and adipose tissue.
Class IA PI3Ks consist of two subunits. The catalytic subunit, p110, contains the kinase domain responsible for the formation of PIP3. The regulatory subunit, the most common of which is p85α, binds to phosphorylated tyrosine residues in tyrosine kinases and their substrate proteins via its SH2 domain, leading to activation of PI3K activity. Both the regulatory and catalytic subunits exist as several different isoforms. In humans, there are four known catalytic subunit isoforms: p110α, p110β, p110δ and p37δ. p110α, p110β and p110δ are encoded by three different genes, PIK3CA, PIK3CB and PIK3CD, respectively (reviewed in 1), whereas p37δ (PIK3CD_ v2) is a splice variant of p110δ3,4. We and others have shown that of these catalytic subunits, p110α is the major contributor for transmitting the insulin signal5–7, whereas p110β becomes active primarily in response to G protein-coupled receptor signaling and plays a role in proliferation8,9. p110δ is more cell specific than p110α and p110β, and plays an important role in immune cells and the embryonic nervous system10–12.
The regulatory subunits are also encoded by three genes, PIK3R1, PIK3R2 and PIK3R3. Their primary gene products are p85α, p85β and p55γ, respectively (reviewed in 1). PIK3R1 also encodes two splice variants of p85α, p55α and p50α, which have more limited tissue distribution. p85α is the major regulatory subunit isoform, constituting 65%–75% of the intracellular pool of regulatory subunits in most cells13. Despite the crucial role of the regulatory subunits of Class IA PI3K in mediating insulin-dependent PI3K signaling14,15, mice with a knockout (KO) of the p85α regulatory subunit display increased insulin sensitivity, increased levels of PIP3 lipids, elevated Akt/PKB activity and improved glucose tolerance13,16–19. The molecular mechanisms that underlie this negative regulation by p85 appear to be complex and include unbalanced stoichiometry between subunits20,21; effects of p85 on both protecting p110 from degradation while partially inhibiting its kinase activity16,19,21,22; retention of PI3K in an inactive vesicle compartment23; links between p85α and PTEN activity24; links between p85α and JNK activity leading to IRS1 serine phosphorylation and inhibition of IRS1-mediated effects, and links between p85α and XBP-1 in modifying the unfolded protein response25.
To dissect the intricate equilibrium between the catalytic and regulatory subunits of PI3K, as well as the opposing and complex roles of p110α and p85α in insulin signaling and action, in the present study, we have investigated the impact of a combined hepatic deletion of p110α and p85α on insulin signaling and whole body glucose homeostasis.
All mice in this study were on a 129Sv-C57Bl/6 mixed genetic background. To create the liver double knock-out mice, p110α lox-lox mice7 were crossed with p85α lox-lox mice, hemizygous for the Albumin-Cre recombinase transgene14. Mice were housed on a 12-hour light cycle and fed a standard rodent chow and water ad libitum. All protocols for animal use and euthanasia were approved by the Gothenburg Ethical Committee on Animal Experiments, in accordance with Swedish guidelines and Directive 2010/63/EU for animal experiments, and by the Animal Care Use Committee of the Joslin Diabetes Center and Harvard Medical School in accordance with National Institutes of Health guidelines. All efforts were made to ameliorate any suffering of the mice by reducing stress, hosting mice in small groups with items that stimulate their natural activity, and allowing the mice to recover 1–2 weeks after each procedure (such as measuring blood glucose, glucose tolerance test etc). During the insulin tolerance test, the mice were monitored closely to not fall too low in blood glucose levels.
For each experiment, a group of 5–12 male mice per genotype were used. The mice were studied from 6 weeks of age to 25 weeks of age. During this time weight and fasting blood glucose levels were measured every two weeks. Glucose tolerance test was performed at 8 weeks, 16 weeks and 24 weeks. Pyruvate tolerance test was performed at 15 weeks and insulin tolerance test was performed at 19 weeks.
Animals were fasted overnight and anesthetized with 2-2-2 tribromoethanol (Sigma-Aldrich, St Louis MO), followed by injection of 5 U of insulin (Actrapid, Novo Nordisk Inc., Plainsboro Township, NJ) or saline via the inferior vena cava. Five minutes after the injection, the liver, muscle and white adipose tissue (WAT) were excised, weighed, and snap-frozen in liquid nitrogen.
Glucose tolerance test was performed by intraperitoneal (i.p.) injection of 2 g glucose/kg BW after an overnight fast. Insulin tolerance test was performed by i.p. injection of 1.25 U insulin/kg BW. Pyruvate tolerance test was performed by i.p. injection of 2 g of pyruvate/kg BW after an over-night fast. Insulin and glucagon was measured with ELISA (Crystal Chem Inc., Downer Grover, IL).
RNA was extracted by homogenization of liver tissue in RLT buffer (Qiagen, Valencia, CA) followed by extraction using the RNeasy kit (Qiagen, Valencia, CA). For gene analysis, cDNA was prepared using a high capacity cDNA archive kit (Applied Biosystems, Foster City, CA) with random hexamer primers. Gene expression was analyzed by real-time reverse transcription-PCR (RT-PCR) on an ABI Prism sequence detection system (Applied Biosystems, Foster City, CA). The cycling conditions used were an initial 95°C 10-minute step followed by 40 cycles of 95°C for 15s and 60°C for 60s. Samples were normalized to the 18S rRNA gene. Primer sequences are available in Table 1.
Liver tissue was homogenized in lysis buffer containing 25 mM Tris-HCl, 2 mM Na3VO4, 10mM Na4P2O7, 1 mM EGTA, 1 mM EDTA, 1% NP-40 and protease inhibitors (Sigma-Aldrich, St Louis MO), then allowed to incubate at 4°C for one hour. Extracts were centrifuged at 55,000 rpm (Beckman 70.1 Ti rotor) for one hour, and the supernatant was stored at -80°C. WAT was homogenized in lysis buffer containing 25 mM Tris-HCl, pH 7.4, 0.5 mM EDTA, 25 mM NaCl, 1% Nonidet P-40, 10 mM NaF, 1 mM orthovanadate, and protease inhibitors (Sigma-Aldrich, St Louis MO) followed by incubation for 2 h at 4°C. The samples were then centrifuged at 12,000 rpm for 15 min, and the supernatant was collected and stored at -80°C. Protein analysis was made by SDS-PAGE and subsequent western blot. Briefly, protein samples were loaded onto 4–12% Bis-Tris protein gels (Thermo-Fisher Scientific, Waltham, MA) and subjected to gel electrophoresis using 25mM Tris, 192 mM glycine and 0.1% SDS as running buffer. Samples were transferred onto a nitrocellulose membrane and the membranes were incubated in 5% skim milk solution for 1 h followed by primary antibody incubation according to the manufacturer’s protocol for each antibody. Membranes were washed 2×5 min and 1×15 min in PBS with 0.1% Tween and then incubated with the secondary antibody for 1h followed by another washing procedure. Immunoprecipitation was performed using magnetic beads coated with protein G (Pierce Biotechnology Inc, Rockford, IL). All western blots and immunoprecipitation experiments were performed with a minimum of four replicates (four separate samples).
IRS1 (RRID:AB_2127860, rabbit monoclonal, 1:50, Cell Signaling Technology Inc, cat# 3407 for western blot), IRS1 (RRID:AB_631842, rabbit polyclonal, 10μl per reaction, Santa Cruz Biotechnology Inc, cat# sc-559 for immunoprecipitation), p110α (rabbit monoclonal, 1:1000 for western blot and 1:50 for immunoprecipitation, Cell Signaling Technology Inc, cat# 4249), p110β (RRID:AB_2165246, rabbit monoclonal, 1:500 for western blot and 1:50 for immunoprecipitation, Cell Signaling Technology Inc, cat# 3011), p110γ (RRID:AB_10828316, rabbit monoclonal, 1:1000, Cell Signaling Technology Inc, cat# 5405), p110δ (mouse monoclonal, 1:500, Becton, Dickinson and Company, cat# 611015), p101 (RRID:AB_10829448, rabbit monoclonal, 1:1000, Cell Signaling Technology Inc, cat# 5569), Vps34 (RRID:AB_2299765, rabbit monoclonal, 1:1000, Cell Signaling Technology Inc, cat# 4263), p150 (rabbit polyclonal, 1:1000, Cell Signaling Technology Inc, cat# 14580), Akt/PKB (RRID:AB_329827, rabbit polyclonal, 1:1000, Cell Signaling Technology Inc, cat# 9272), pS-Akt/PKB (RRID:AB_329825, rabbit polyclonal, 1:1000, Cell Signaling Technology Inc, cat# 9271), pT-Akt/PKB (RRID:AB_2255933, rabbit monoclonal, 1:1000, Cell Signaling Technology Inc, cat# 2965), pT-p70S6K (RRID:AB_330944, rabbit polyclonal, 1:1000, Cell Signaling Technology Inc, cat# 9205), p70S6K (rabbit polyclonal, 1:500, Cell Signaling Technology Inc, cat# 9202), p85α (RRID:AB_2268174, rabbit monoclonal, 1:1000, Abcam, cat# 22653), p85-pan (RRID:AB_10831521, rabbit monoclonal, 1:1000, Cell Signaling Technology Inc, cat# 4257), pT202/Y204-ERK (RRID:AB_2315112, rabbit monoclonal, 1:1000, Cell Signaling Technology Inc, cat# 4370), ERK (RRID:AB_390779, rabbit monoclonal, 1:1000, Cell Signaling Technology Inc, cat# 4695), p55γ (mouse monoclonal, 1:2000, Abcam, cat# ab186612), PIK3C2α (rabbit polyclonal, 1:1000, MyBiosource, cat# MBS9202698), PIK3C2γ (rabbit polyclonal, 1:1000, MyBiosource, cat# MBS820611). Rabbit secondary antibody (RRID:AB_772206, HRP-linked from donkey, 1:1000, GE Healthcare Life Sciences, cat# NA934), mouse secondary antibody (RRID:AB_772210, HRP-linked from sheep, 1:1000, GE Healthcare Life Sciences, cat# NA931).
Immunoprecipitates, using protein G Dynabeads (Life Technologies), of liver protein lysates were prepared with p110α and p110β antibodies or IRS1 antibody. The PI3K assay was performed as previously described13. Briefly, immunoprecipitates were incubated with 5 μg PI substrate (phosphatidylinositol from bovine liver), 20 mM MgCl 2, 8 μM cold ATP and 0.5 μl radio-labeled [γ- 32P]-ATP (1.11*1014 bq/mmol) in PI3K reaction buffer (20 mM Tris-HCl, 100 mM NaCl and 0.5 mM EGTA) for 25 min at room temperature. The resulting radioactively labeled PIP was analyzed with thin layer chromatography and phosphorimaging (FLA-3000, Fujifilm). Prior to these experiments, titration experiments of bead concentration and PI substrate concentration were performed to ensure precipitation of equal amounts of protein, as well as optimal PI concentration to obtain maximal enzyme activity.
Mice with a liver-specific deletion of p110α and p85α, termed hereafter liver double knockout (L-DKO) mice, were created by breeding mice carrying homozygous floxed Pik3ca and Pik3r1 alleles14,26 with transgenic mice carrying the Cre recombinase driven by the albumin promoter (albumin-Cre). Deletion of Pik3ca and Pik3r1 in the liver resulted in markedly reduced gene and protein expression of p110α and p85α (Figures 1A, 1C, 1E), as well as impaired activation of the downstream targets Akt/PKB, with decreased phosphorylation of serine 473 and threonine 308, and p70S6 kinase (Figure 1F–I). p110β gene expression was not affected by the deletion of p110α and p85α (Figure 1B). p85β gene expression was slightly, but significantly, decreased in the L-DKO livers (Figure 1D). As expected, in the floxed control mice, there was an increase in the amount of p110α associated with IRS1 in response to insulin compared to basal conditions, whereas no p110α was associated with IRS1 in the L-DKO mice (Figure 1K and 1L). MAPK signaling, as shown by ERK phosphorylation, was unchanged in the L-DKO mice compared to controls (Figure 1F and 1J).
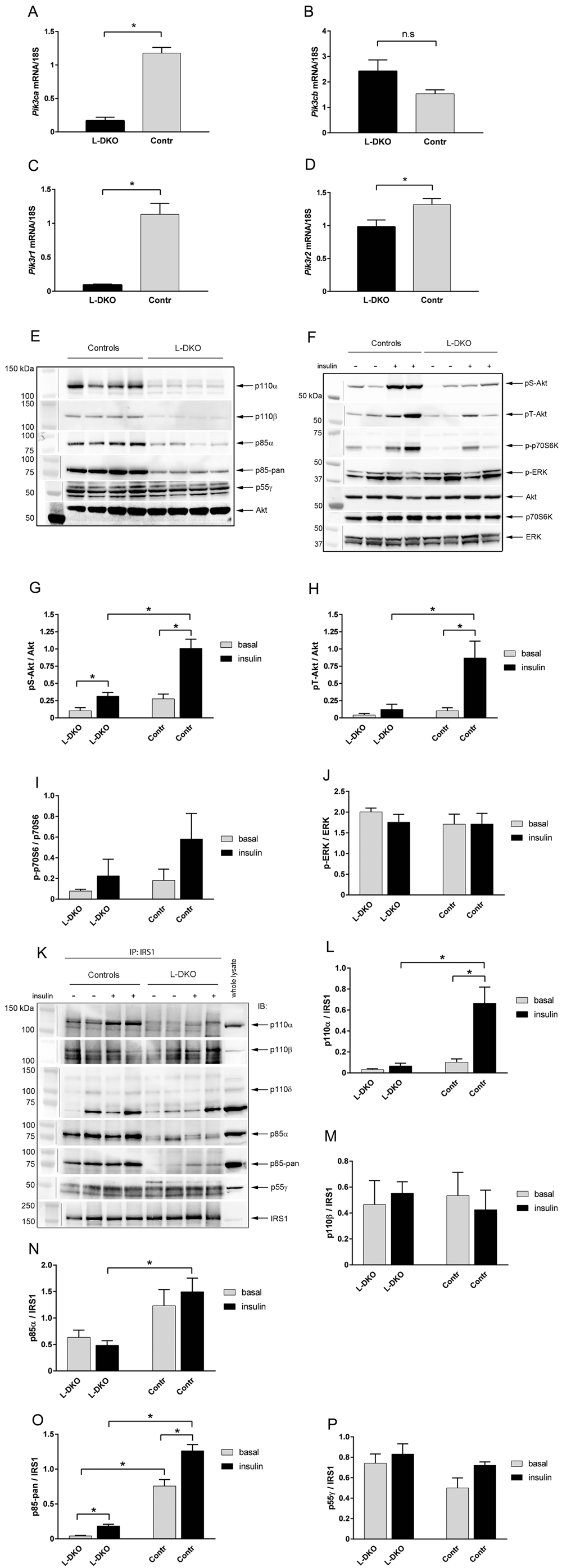
mRNA expression of (A) Pik3ca, (B) Pik3cb, (C) Pik3r1 and (D) Pik3r2 in livers of flox controls and L-DKO mice. (E) Representative western blot of protein expression of p110α, p110β, p85α, p85-pan (detects both p85 isoforms) and p55γ. Total Akt was used as a loading control. (F, phosphorylated Akt/PKB (Ser 473 and Thr 308), phosphorylated p70S6 kinase, and phosphorylated ERK in livers of flox controls and L-DKO mice. Total Akt, p70S6K and ERK was used as loading controls. (G–J) Quantification measurements of the western blots shown in (F) of pS-Akt, pT-Akt, p-p70S6K and p-ERK respectively. (K) Representative western blot of immunoprecipitation experiments with antibodies for IRS1 and subsequent immunoblotting with antibodies for p110α, p110β, p110δ, p85α, p85-pan (detects both p85 isoforms) and p55γ. The whole-lysate reference sample was from an insulin-treated flox control mouse. (L–P) Quantification measurements of the western blots shown in (K) of p110α, p110β, p85α, p85-pan and p55γ. IP = immunoprecipitation, IB = immunoblot. Basal condition is indicated with a minus (-) sign, insulin-treated condition is indicated with a plus (+) sign. Basal conditions refer to fasting of mice overnight and injection of saline through the vena cava. Insulin treatment refers to injection of 5 U of insulin through the vena cava 5 min prior to euthanization. Error bars indicate SEM (n = 5–8). *, p < 0.05 compared to controls. n.s = non significant. Images containing a vertical line are composites taken from a single original image.
Previous studies have shown that the interaction between the regulatory and catalytic subunits of PI3K to form dimers has a mutual stabilizing effect on both subunits, whereas the monomeric forms are more readily subjected to degradation19,21,22. We hypothesized that more p85β would bind to IRS1 when p85α was absent, thereby maintaining p110β stabilization. However, only very low levels of p85β protein were detected in the liver of the L-DKO mice, as shown both in assessment of total p85 protein (Figure 1E) and in p85 immunoprecipitates with IRS1 (Figure 1K and 1O), similar to what we have reported earlier in liver-specific p85α knock-out mice14. The expression of p55γ regulatory isoform protein was not affected by deletion of p110α and p85α (Figure 1E), nor was the amount bound to IRS1 (Figure 1K and 1P). In contrast, total p110β protein expression was decreased in the L-DKO mice compared to controls (Figure 1E), likely due to destabilization of this catalytic isoform in the absence of p85α, supporting an insufficiency for p85β to compensate for the loss of p85α. Interestingly, despite overall decreased protein expression of p110β, similar amounts of p110β were associated with IRS1 in controls and L-DKO mice (Figure 1K and 1M). The third catalytic isoform of class IA PI3Ks, p110δ, has been shown to have a major role in immune cells and the embryonic nervous system, but not in other tissues10–12. Consistent with this, we found only very small amounts of full length p110δ in whole liver lysates or bound to IRS1 (Figure 1K). However, the antibody picked up a band of about 70 kDa in whole lysates and in IRS1 immunoprecipitates (Figure 1K). The amount of this protein did not appear to be consistently different between controls and L-DKO mice or between basal and insulin-stimulated samples, so is likely non-specific.
As expected, p110α kinase activity, as assessed by the ability to add the 3’-phosphate group to phosphoinositides in p110α immunoprecipitates, was markedly decreased both in the basal- and insulin-stimulated states of the L-DKO mice compared to controls (Figure 2A). Overall p110β kinase activity was much lower than p110α kinase activity in control mice, consistent with previous studies7,27, and was not changed in the basal state between controls and L-DKO mice (Figure 2B). However, p110β kinase activity was significantly decreased in the insulin-stimulated state of the L-DKO mice (Figure 2B), even though similar amounts of p110β protein were associated with IRS1 in controls and L-DKO mice (Figure 1K and 1M). Surprisingly, IRS1-associated phosphatidylinositol kinase activity in response to insulin was intact in the L-DKO, despite lack of p110α kinase activity and decreased p110β activity (Figure 2C).
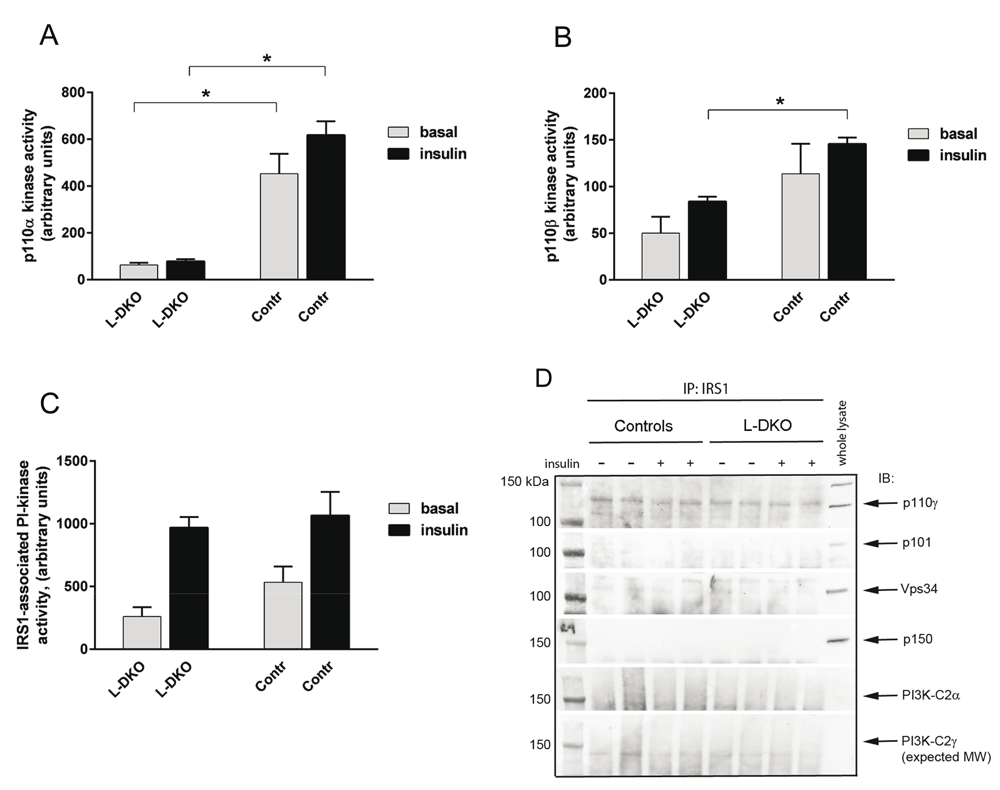
(A) p110α lipid kinase activity, (B) p110β lipid kinase activity, and (C) IRS1-associated phosphatidylinositol (PI) kinase activity in L-DKO mice and controls were assessed by immunoprecipitation. (D) Representative western blot of immunoprecipitation experiments with antibodies for IRS1 and subsequent immunoblotting with antibodies for p110γ, p101, Vps34, p150, PIK3-C2α and PIK3-C2γ. The whole-lysate reference sample was from an insulin-treated flox control mouse. IP = immunoprecipitation, IB = immunoblot. Basal condition is indicated with a minus (-) sign, insulin-treated condition is indicated with a plus (+) sign. Basal conditions refer to fasting of mice overnight and injection of saline through the vena cava. Insulin treatment refers to injection of 5 U of insulin through the vena cava 5 min prior to euthanization. Error bars indicate SEM (n = 4). *, p < 0.05 compared to controls.
To explore if other classes of phosphoinositide 3-kinases were responsible for the intact IRS1-associated activity, we investigated the association of IRS1 with class IB-, class II and class III members of phosphoinositide 3-kinases in liver lysates from controls and L-DKO mice. Class IB PI3K consists of the p110γ catalytic subunit and the p101 regulatory subunit. The amount of p101 was low in whole lysates and no p101 was associated with IRS1 in either control mice or L-DKO mice (Figure 2D). p110γ was associated with IRS1, but the amounts were similar between controls and L-DKO mice (Figure 2D). Class III PI3K catalytic subunit Vps34 and regulatory subunit p150 were present in whole lysates, but not associated with IRS1 in either control mice or L-DKO mice (Figure 2D). Class II PI3K consists of only one subunit, but exists in several isoforms. The PI3K-C2α isoform is ubiquitously expressed, whereas PI3K-C2γ has been reported to have a more limited tissue distribution, including hepatocytes28,29. However, we detected only little or no PI3K-C2α or PI3K-C2γ in whole liver lysates or associated with IRS1 in controls or L-DKO mice (Figure 2D). Thus, other classes of phosphoinositide 3-kinases did not compensate for the loss of p110α and p85α in the L-DKO mice and could not explain the intact IRS1-associated phosphatidylinositol kinase activity in the absence of p110α and p85α.
On standard chow (4% fat content by weight, 12% by calories), L-DKO mice had similar body weights to the flox control mice until 10–12 weeks of age, after which the L-DKO mice showed slower weight gain compared to controls (Figure 3A). At least part of this decrease was due to a decrease in liver weight. Thus, by 10 weeks of age, the ratio of liver weight to body weight in the L-DKO mice was decreased by 13% compared to control mice (Figure 3B), whereas there was no difference in WAT weight or muscle weight (Figures 3C and 3D). At 24 weeks of age, liver weight remained decreased (18%), and there was also a significant decreased WAT weight (22%) for the L-DKO mice compared to controls (Figures 3B and 3C). To assess a possible change in insulin sensitivity in WAT, associated with the decreased WAT mass, we investigated insulin signaling mediators in this tissue. WAT p110α- and p85α expression was similar in controls and L-DKO mice as was Akt/PKB phosphorylation in response to insulin (Figure 3E). There was no difference in insulin-stimulated Akt/PKB-activation at 10w compared to 25w in either controls or L-DKO mice (Figure 3E).
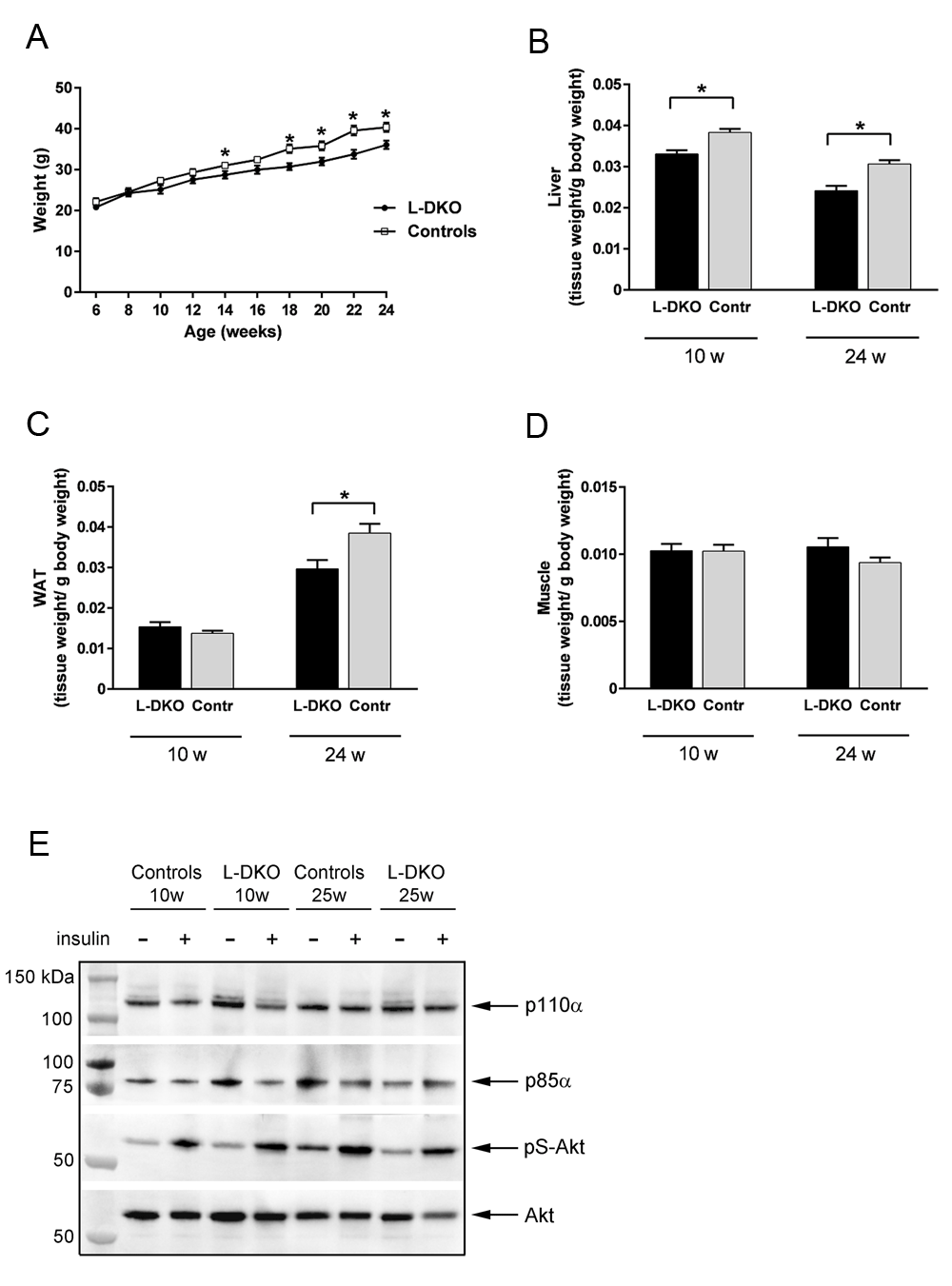
(A) whole body weight, (B) liver weight, (C) white adipose tissue (WAT) weight and (D) muscle weight of 10 and 24 week old male L-DKO mice and controls. (E) Representative western blot of p110α, p85α and phosphorylated Akt/PKB (Ser 473) in WAT of 10 and 25 week old male flox controls and L-DKO mice. Total Akt/PKB was used as a loading control. Basal condition is indicated with a minus (-) sign, insulin-treated condition is indicated with a plus (+) sign. Basal conditions refer to fasting of mice overnight and injection of saline through the vena cava. Insulin treatment refers to injection of 5 U of insulin through the vena cava 5 min prior to euthanization. Error bars indicate SEM (n = 8–17). *, p < 0.05 compared to controls. w = weeks.
Despite similar body weight (Figure 3A), L-DKO mice were severely glucose intolerant as early as at 8 weeks of age (Figure 4A), and remained similarly glucose intolerant throughout the 24 week study (Figures 4B and 4C). This was associated with markedly increased fasting insulin levels (Figure 4D). However, somewhat surprisingly, the L-DKO mice showed a normal response to exogenous insulin during an intraperitoneal insulin tolerance test (Figure 4E). Despite the markedly impaired glucose tolerance and marked hyperinsulinemia, fasting and random fed glucose levels in the L-DKO mice remained similar between controls and L-DKO mice (Figures 4F and 4G). Circulating glucagon levels were also similar between L-DKO mice and controls both at 10 weeks of age and 24 weeks of age (Figure 4H).
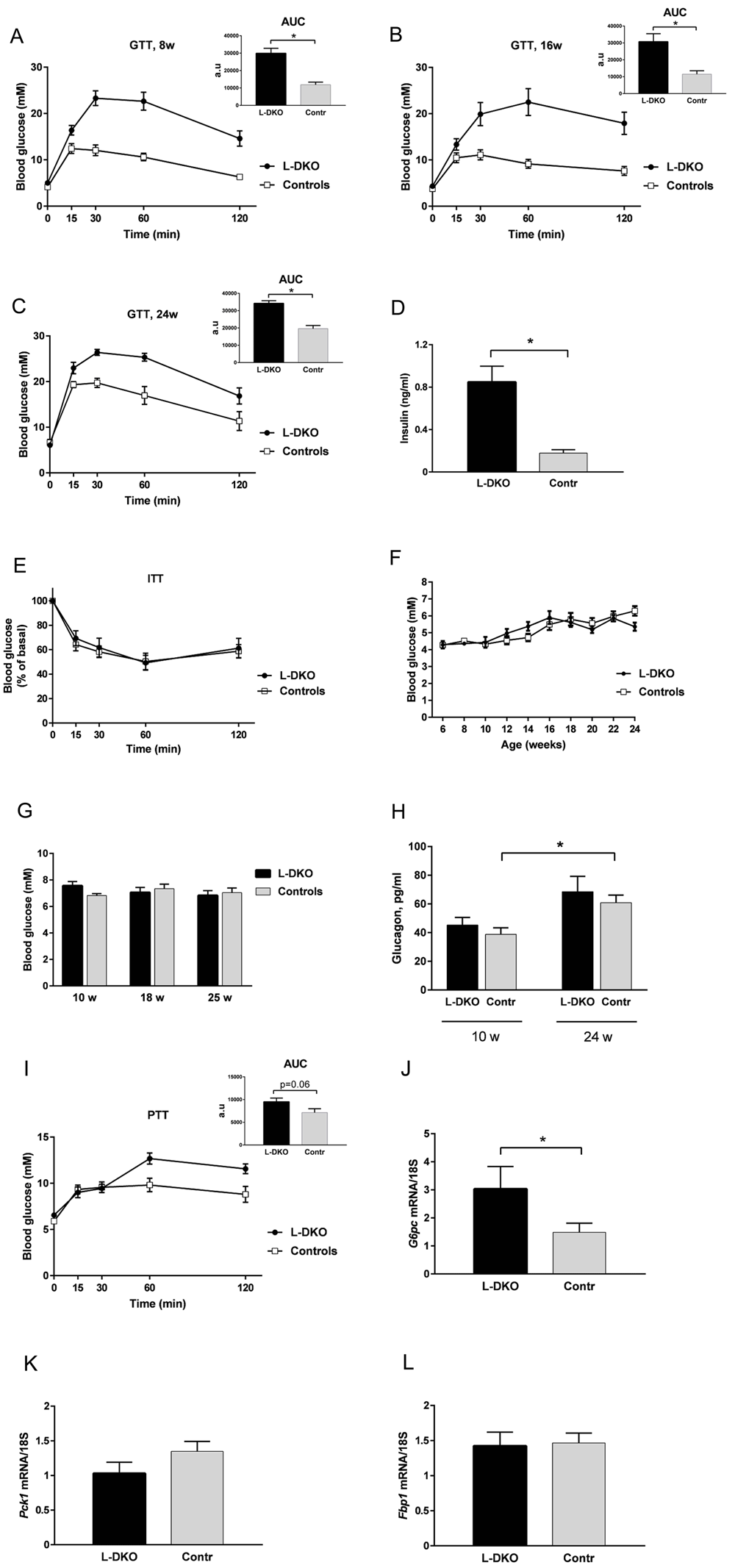
Glucose tolerance test at (A) 8 weeks, (B) 16 weeks and (C) 24 weeks of age; (D) fasting insulin levels at 10 weeks of age; (E) insulin tolerance test at 19 weeks of age; (F) fasting glucose levels; (G) random fed glucose levels; (H) fasting glucagon levels; (I) pyruvate tolerance test at 15 weeks of age; (J–L) hepatic mRNA expression of the gluconeogenic markers glucose 6-phosphatase (G6pc), phosphoenolpyruvate carboxykinase (Pck1) or fructose 1,6-bisphosphatase (Fbp1). L-DKO mice and controls were given 2 g glucose/kg body weight intraperitoneally (i.p.) for the glucose tolerance test or 1.25 U insulin/kg body weight i.p. for the insulin tolerance test or 2 g pyruvate/kg body weight i.p. for the pyruvate tolerance test. Blood glucose was measured at 0, 15, 30, 60, and 120 min. Error bars indicate SEM (n =6–12). *, p < 0.05. The insets in the glucose tolerance test graphs and the pyruvate tolerance test graph show the area under the curve (AUC) with subtracted basal glucose values.
Increased gluconeogenesis is one of the hallmarks of hepatic insulin resistance in type 2 diabetes. To determine whether the impaired glucose tolerance in L-DKO mice was due to increased hepatic glucose output, we subjected these animals to a challenge with pyruvate, the major gluconeogenic substrate. Over the 120 min period following administration of pyruvate, there was a trend toward increased glucose levels in L-DKO mice compared to controls, but this was not statistically significant (Figure 4I). This was associated with a significant change in hepatic gene expression of the gluconeogenic enzyme glucose 6-phosphatase (G6pc) (Figure 4J), whereas gene expression of the other key mediators of gluconeogenesis, phosphoenolpyruvate carboxykinase (Pck1) and fructose 1,6-bisphosphatase (Fbp1), remained similar between control mice and L-DKO mice (Figures 4K and 4L).
In this study, we investigated the impact of a combined deletion of p110α and p85α on insulin signaling and glucose homeostasis. For this purpose, we created mice with a liver-specific deletion of the major catalytic and major regulatory subunits of PI3K: Pik3ca and Pik3r1. We have previously shown that hepatic deletion of only p110α results in severe insulin resistance and impaired glucose tolerance, signifying that p110α is crucial for mediating insulin signaling7. Moreover, mice deficient in all p85 isoforms in either muscle or liver exhibit severely impaired insulin signaling in these tissues14,15. The liver plays a crucial role in maintaining glucose homeostasis; we therefore hypothesized that deleting both these isoforms would result in severe and overt diabetes.
As expected, in the L-DKO mice, p110α catalytic activity was blunted and, as a result, the activation of the signal downstream of PI3K was markedly decreased. However, the L-DKO mice showed normal fasting and fed blood glucose levels throughout the study (24 weeks) and normal insulin tolerance. Glucose tolerance was impaired in the L-DKO mice and circulating insulin levels were markedly elevated, but to a degree similar to mice with only p110α deleted in the liver (L-p110α KO)7. Surprisingly, despite abolished p110α activity, we observed an intact total IRS1-associated phosphatidylinositol (PI) kinase activity in the L-DKO mice. This finding was very different from what we previously observed in L-p110α KO mice, which only have p110α deleted in the liver7. In the L-p110α KO mice, insulin-stimulated IRS1-associated PI kinase activity was markedly blunted7. This suggests that it is the loss of the regulatory subunit that accounted for the preserved IRS1-associated activation, possibly by enabling other phosphatidylinositol kinases to bind to IRS1. However, there did not appear to be any compensatory effects of other known catalytic and regulatory class IA subunit isoforms for the action of insulin. Thus, we did not detect any differences in the IRS1-associated amounts of p110β, p110δ or p55γ in controls and L-DKO mice in response to insulin and only very small amounts of p85β associated with IRS1 in the L-DKO mice. Similarly, no evidence of compensatory effects by class IB, class II or class III members of phosphoinositide 3-kinases were found.
Previous studies by us and others, including our study of the L-p110α KO mice, have shown that p110β is unable to compensate for the loss of p110α7,26,30. However, we observed similar amounts of IRS1-associated p110β in the L-DKO mice and controls despite overall total decreased levels of p110β in the liver. We therefore speculated that perhaps p110β activity was increased in response to insulin in the L-DKO mice compared to controls, which would explain the sustained IRS1-associated PI kinase activity. We found no difference in the p110β activity between the controls and the L-DKO mice in the basal state. In addition, the p110β kinase activity was significantly decreased, rather than increased, in the insulin-stimulated state of L-DKO mice. We thus conclude that the sustained IRS1-associated PI kinase activity in the L-DKO mice is not due to increased activity of p110β.
Although the amount of IRS1-associated p110β was similar in controls and L-DKO, the total amount of p110β was decreased in the L-DKO mice. We, and others, have previously reported that the dimeric interaction between the regulatory and the catalytic subunits results in stabilization of the subunits, whereas the monomeric forms are more readily subjected to degradation18,20–21. Therefore, the absence of the major regulatory subunit p85α in the L-DKO mice probably subjects p110β to more rapid degradation. In this context, observing similar amounts of p110β associated with IRS1 in controls and L-DKO mice is somewhat surprising, but is likely due to stabilization of a fraction of p110β subunits by interaction with IRS1-associated p85β or p55γ.
Lack of the major regulatory subunit in the L-DKO mice, accompanied by absence of increased activation of p110β or compensatory increased expression of other phosphoinositide 3-kinases, suggests that presence of other classes of phosphatidylinositol kinases, perhaps PI4K and PI5K, account for the intact IRS1-associated kinase activity by directly binding to IRS1. PI4Ks have been described as mediators of endosomal trafficking from the Golgi and to be involved in EGF-stimulated phosphoinositide signaling31. Type II PI4Ks interact with the EGF receptor, but they are not known to interact with IRS1. Type III PI4Ks are structurally related to PI3Ks, with a high degree of conservation between their catalytic domains and sensitivity to wortmannin31. The isoform PI4KIIIα has been reported to be functionally connected to PI3K in FGF signaling during pectoral fin development in the zebra fish32.
PI5K exists as two separate classes, PI(3)P5K and PI(4)P5K, phosphorylating the D5 position of the inositol ring of phosphatidylinositol 3-phosphate and phosphatidylinositol 4-phosphate, respectively. Of the PI(4)P5Ks, the isozyme PIP5Kc has been shown to respond to, and become phosphorylated by, various hormones and growth factors, such as EGF, and play a role in actin cytoskeletal reorganization, clathrin-dependent endocytosis, membrane ruffle formation, etc.33. However, a direct effect on insulin signaling and interaction with IRSs by PIP5Kc has not been reported. PI(3)P5K, also called PIKfyve, and has been quite extensively studied and reported to be involved in membrane trafficking, stress- or hormone-induced signaling, ion channel activity, cytoskeletal dynamics, nuclear transport, gene transcription and cell cycle progression34. Interestingly, PIKfyve is regulated by insulin, recruiting PIKfyve to inner membranes, where insulin receptor and IRSs are also found35, and co-precipitates with p110 and p85 subunits in 3T3-L1 adipocytes36. Thus, PIKfyve appears to be a possible contributor to the sustained IRS1-associated kinase activity in the L-DKO mice. However, PIKfyve expression has been reported to be rather tissue specific, mainly expressed in adipose tissue, muscle and brain37,38 and expression in the liver appears low37,38. A more extensive follow-up investigation of the various phosphoinositides in the L-DKO livers compared to controls may help elucidating the phosphatidylinositol kinase responsible for the sustained IRS1-associated kinase activity in the L-DKO mice.
The absence of both p85α and p110α, as seen in the L-DKO mice, would logically result in a very severely impaired metabolic phenotype. The L-DKO mice had markedly elevated circulating insulin levels, impaired glucose tolerance and showed a trend toward increased rates of hepatic glucose output when given pyruvate. However, fasted and fed glucose levels were not different between controls and L-DKO mice, i.e., randomly fed L-DKO mice were not diabetic and insulin tolerance tests were normal. Part of this protection might be the fact that L-DKO mice had less accumulation of WAT with age. In addition, it is possible that the high circulating insulin levels reflect an impaired hepatic insulin clearance rather than insulin resistance in the muscle, which would explain the paradoxical normal insulin tolerance. Interestingly, body weight, WAT weight and hepatic glucose output were significantly increased and insulin tolerance severely impaired in L-p110α KO mice7, which lack only p110α in liver, demonstrating that L-DKO mice showed an overall less severe metabolic phenotype compared to L-p110α KO mice.
In summary, deletion of hepatic p110α and p85α results in an impaired insulin signal and impaired glucose homeostasis, but shows an overall less severe metabolic phenotype compared to mice with only p110α deleted in the liver. Although other PI3Ks were unable to compensate for the loss of p110α and p85α, IRS1-associated phosphatidylinositol kinase activity was surprisingly still intact, possibly due to interaction of IRS1 with other classes of phosphatidylinositol kinases.
Abbreviations: Fbp, fructose-1,6-bisphosphatase; G6pc, glucose-6-phosphatase; GTT, glucose tolerance test; ITT, insulin tolerance test; L-DKO, liver double knockout; Pck, phosphoenolpyruvate carboxykinase; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PKB, protein kinase B; PTT, pyruvate tolerance test; WAT, white adipose tissue
Dataset 1: Raw data for gene and protein expression shown in Figure 1. DOI, 10.5256/f1000research.12418.d20590939
Dataset 2: Raw data for PI3K activity measurements and protein expression shown in Figure 2. DOI, 10.5256/f1000research.12418.d17549440
Dataset 3: Raw data for body and tissue weights and protein expression shown in Figure 3. DOI, 10.5256/f1000research.12418.d17549541
Dataset 4: Raw data for metabolic procedures and measurements and gene expression shown in Figure 4. DOI, 10.5256/f1000research.12418.d20591042
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)