Keywords
Genomics, Plasmodium falciparum, microarray, Hidden markov model, clustering, Functional Enrichment.
This article is included in the Pathogens gateway.
Genomics, Plasmodium falciparum, microarray, Hidden markov model, clustering, Functional Enrichment.
We have responded and implemented detailed explanations of the methodology, and the Go Term enriched (p-value) has been provided. The normalized and discretized data sets used have been provided as Dataset 1.
Technological advancement in bioinformatics such as the high through put sequencing technology has resulted in the availability of a very large amount of informative data1. Expressions of thousands of genes are now being measured concurrently under various experimental conditions using microarray technology. Microarray consists of many thousands of short, single stranded sequences, each immobilized as individual elements on a solid support, that are complementary to the cDNA strand representing a single gene. Gene expression measurements can be obtained for thousands of genes simultaneously using microarray technology. In a cell, genes are transcribed into mRNA molecules which in turn can be translated into proteins, and it is these proteins that perform biological functions, give cellular structure and in turn regulate gene expression2.
Various researches had been carried out on the analysis and extraction of useful biological information such as detection of differential expression, clustering and predicting sample characteristics3. One of the important of gene expression data is the ability to infer biological function from genes with similar expression patterns4. Due to large number of genes and complexity of the data and networks, research has suggested clustering to be a very useful and appropriate technique for the analysis of gene expression data which can be used to determine gene coregulation5, subpopulation6, cellular processes, gene function7 and understanding disease processes8.
Clustering algorithms as described by 9 can either be distance based or model based. Distance-based clustering such as the k means10 does not consider dependencies between time points while the model based approach embeds time dependencies and uses statistical models to cluster data. Other approaches include Self Organizing Maps11, Principal Component Analysis, hierarchical clustering4, graph theory approach12, genetic algorithms and the Support Vector Machine13. These algorithms has been successfully applied to various time series data but still have various shortcoming such as determining the optimal number of clusters and choosing the best algorithm for clustering since most of them are based on heuristics. The algorithms are also not very effective especially when a particular gene is associated with different clusters and performs multiple functions.
This research focuses on clustering of genes with similar expression patterns using Hidden Markov Models (HMM) for time course data because they are able to model the temporal and stochastic nature of the data. A Markov process is a stochastic process such that the state at every time belongs to a finite set, the evolution occurs in a discrete time and the probability distribution of a state at a given time is explicitly dependent only on the last state and not on all the others. This refers to as first-order Markov process (Markov chain) for which the probability distribution of a state at a given time is explicitly dependent only on the previous state and not on all the others. That is, the probability of the next (“future”) state is directly dependent only on the present state and the preceding (“past”) states are irrelevant once the present state is given14. Fonzo et al.14 defined HMM as a generalization of a Markov chain in which each (“internal”) state is not directly observable (hidden) but produces (“emits”) an observable random output (“external”) state, also called “emission” state. Schliep et al.9 proposed a partially supervised clustering method to account for horizontal dependencies along the time axis and to cope with the missing values and noise in time course data. This approach used k means algorithm and the Baum welch algorithm for parameter estimation. Further analysis on the cluster was done with the Viterbi algorithm which gives a fine grain, homogeneous decomposition of the clusters. The partial supervised learning was done by adding labeled data to the initial collection of clusters. Ji et al.15 developed an application to cluster and mine useful biological information from gene expression data. The dataset was first normalized to a mean of zero and variance of one and then discretized into expression fluctuation symbol with each symbol representing either an increase, decrease or no change in the expression measurement. A simple HMM was constructed for these fluctuation sequences. The model was trained using the Baum-Welch EM algorithm and the probability of a sequence given a HMM was calculated using the forward-backward algorithm. Several copies of the HMM was made so that each copy represent a single cluster. These clusters were made to specialize by using a weighted Baum-Welch algorithm where the weight is proportional to the probability of the sequence given the model. The Rand Index and Figure of Merit was used to validate the optimal number of cluster results.
Geng et al.16 developed a gene function prediction tool based on HMM where they studied yeast function classes who had sufficient number of open reading frame (ORF) in Munich Information Center for Protein Sequences (MIPS). Each class was labeled as a distinct HMM. The process was performed on three stages; the discretization, training and inference stages and data used for this analysis was the yeast time series expression data. Lees et al.17 proposed another methodology to cluster gene expression data using HMM and transcription factor information to remove noise and reduce bias clustering. A single HMM was designed for the entire data set to see if it would affect clustering results. Each path in the HMM represents a cluster, transition between states in a path is set to a probability of 1 and transition between states on different path is set to a probability of 0. Genes were allocated to clusters by calculating the probability of each sequence produced by the HMM. Clusters were validated using the likelihood ratio test which computes the difference in the log-likelihood of a complex model to that of a simpler model. Zeng and Garcia-Frias18 implemented the profile HMM as a self-organizing map, this profile HMM is a special case of the left to right inhomogeneous HMM which is able to model the temporal nature of the data. This makes it very useful for real life applications. The profile HMM is trained using the Baum-Welch algorithm19 and clustering was done using the Viterbi algorithm and the algorithm was implemented on the fibroblast and the sporulation datasets. Beal et al.20 implemented the Hierarchical Dirichlet Process Hidden Markov Model (HDP-HMM) for clustering gene expression data with countably infinite HMM. Gibbs sampling method was used to reduce the time complexity of the inference. The data used for the implementation was derived from Lyer et al.21 and Cho et al.22 which was normalized and standardized to a log ratio of 1 at time t = 1. Baum Welch algorithm was used for Estimation Maximization. In this work, we adopted the work of Lee et al.17 and applied HMM on the Plasmodium falciparum RNA-seq dataset.
In this work, data was extracted and normalized to a mean of 1 and standard deviation of 0. Discretization was done on the data to improve the clustering results. The HMM forward-backward algorithm and Baum-Welch training algorithm was implemented to cluster the gene expression data. Genes were then assigned to cluster using the forward algorithm and inference was done by obtaining functionally enriched genes in the cluster set using FunRich tool. The data used was published by 23, they used the Illumina based sequencing technology to extract expressions of 5270 P. falciparum genes at seven different time points every 8hrs for 42hrs. The clustering algorithms was implemented by first randomly initializing all the HMM parameters, then, forward algorithm was implemented to calculate the forward probabilities of the observation and Baum-Welch algorithm was used for data fitting, then the likelihood of each HMM was calculated iteratively until the optimal likelihood is obtained. This process is repeated for all the different sized HMMs used.
HMMs can be viewed as probabilistic functions of a Markov chain24 such that each state can produce emissions according to emission probabilities.
Definition 1. (Hidden Markov Model). Let O = (O1,…) be a sequence over an alphabet ∈. A Hidden Markov Model λ is determined by the following parameters:
• Si, the states i = 1, … N
• πi, the probability of starting in state Si,
• αij, the transition probability from state Si to state Sj, and
• bi(ω), the emission probability function of a symbol ω ∈ Σ in state Si.
Definition 2. (Hidden Markov Cluster Problem). Given a set O = {O1, O2,…, On} of n sequences, not necessarily of equal length, and a fixed integer K ≪ n. Compute a partition C = (C1, C2,…, Ck) of O and HMMs λ1,…, λk as to maximize the objective function
Where L(Oi | λk) denotes the likelihood function for generating sequence Oi by model λk:
The preprocessing was done in two stages. The first stage was the normalization and the second stage was discretization. The normalization was done with the R statistical package using the normalize library. Normalization removes static variation in the microarray experiment which affects the gene expression level. Normalization also helps in speeding up the learning phase. Missing values are also removed during normalization. The data used after normalization is given in Data Availability as normalized.xsl. Discretization was done by converting the time points to symbols depending on whether the expression value has increased, decreased or not changed, as given in Data Availability as Discretized_Data.txt. This is done by using the equation below.
Where:
Si = fluctuation level between time i and i + 1
Ei = expression level at time point i
L = number of time points.
a = threshold value between timepoints.
In this work, we implemented a model-based HMM clustering algorithm where a cluster represents a path in the HMM model. Therefore, as the number of cluster increases, the number of paths through the model increases and the HMM becomes larger and larger. The number of hidden state is the number of clusters multiplied by the sequence length. Research has also shown that HMM that transverse from left to right best models time series data, therefore HMM moves from only right-to-left. The structure of the HMM is shown in Figure 1.
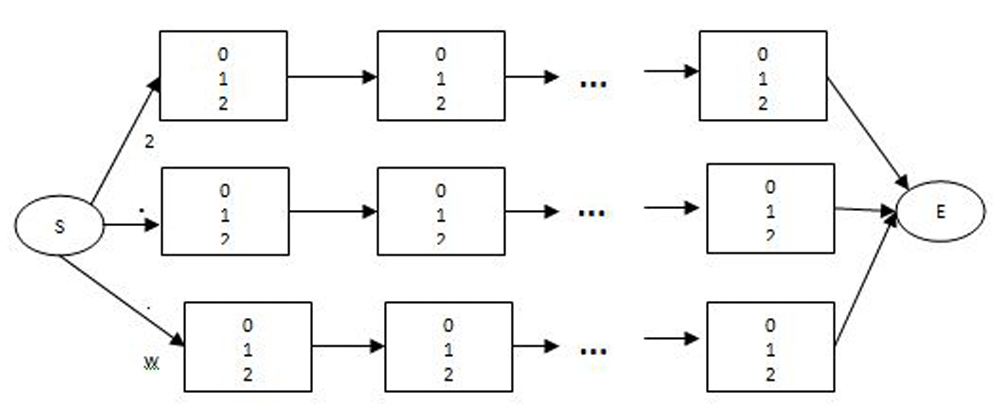
The number 0, 1, and 2 represents the emmission symbols at each state.
For an HMM to be used for real world applications, there are three basic problems of interest that needs to be solved.
1. Given an observation sequence O = {O1 … On}, and a model λ = (A, B, π), how can we effectively compute the probability of observation given the model, i.e P(O/λ)
2. Given an observation sequence and a model λ, how do we choose a state sequence which best explains the observation.
3. How do we adjust the model parameter to optimize P(O/λ).
Problem 1 is the evaluation problem and solution allows us to choose the model that best matches the observation. This can be solved using the forward-backward algorithm. Problem 2 aims to discover the hidden state in the model and find correct state for each observation. It is also known as the inference problem. There is no correct solution for this problem but an optimal solution can be reached based on a stated optimality criterion. This problem can be solved using the Viterbi algorithm. The third and most difficult problem aims to adjust the model parameter to optimize the HMM and is also known as training the HMM. It is usually solved using the Baum-Welch training algorithm. In this work, we implemented the HMM clustering algorithm in C++ programming language. HMM structure for clustering was first created. A cluster is represented as a path in the HMM, for example, for x cluster the model increases proportionally with x paths through the HMM. The prior, transition and observation probability are then estimated for each cluster size by using the solution to problem 3 to optimize each model. Then, genes are assigned to each cluster by choosing the best model/cluster that fits an observation using the forward algorithm.
The left to right model has the property that, as the time increases, the state transition increases, that is, the states moves from left to right. This process is conveniently able to model data whose properties change over time.
The forward algorithm calculates the forward probabilities of a sequence of observation. This is the probability of getting a series of observation given that the HMM parameters (A, B, π) are known. This computation is usually expensive computationally. The time invariance of the probabilities can however be used to reduce the complexity of this algorithm by calculating the probabilities recursively. These probabilities are calculated by computing the partial probabilities for each state from times t = 1 to t = T. The sum of all the final probabilities for each states is the probability of observing the sequence given the HMM and this was used in this research to compute the likelihood of a sequence given the HMM. The algorithm is in 3 stages and illustrated as follows:
Initialization stage
This initializes the forward probability, which is the joint probability of starting at state and initially observing O1.
Induction stage
This is the joint probability of observation and state 3 at time t + 1 via state at time t (i.e. the joint probability of observing o at state 3 at time t+1 and form state and time t). This is performed at all states and is iterated for all times from t = 1 to T-1.
Termination stage
This computes all the forward variables, which is the P(o|M).
Where:
αij = transition from state i to j
bi(o) = probability of emitting a symbol in a particular state
t = time
M = model
αi = forward variable of state i
π = probability of starting at a particular state
The backward algorithm like the forward algorithm calculates backward probabilities instead. The backward probability is the probability of starting in a state at a time t and generating the rest of the observation sequence Ot + 1,…, OT. The backward probability can be calculated by using a variant of the forward algorithm. Therefore, instead of starting at time t = 1, the algorithm starts at time t = T and moves backwards from OT to Ot + 1. In this work, the backward algorithm was used alongside the forward algorithm to re-estimate the HMM parameters. This algorithm also involves three steps and is illustrated below.
Initialization
This is the initial probability of being in a state Si at time T and generating nothing. The value of this computation is usually 1.
Induction
This step calculates the probabilities of partial sequence observation from t+1 to end given state at time t and the model λ.
Termination
It calculates all the backward variables which is the P(Ot+1, …, OT|Q1 = Si).
Where:
αij = transition from state i to j
bj(o) = probability of emitting a symbol in a particular
βt(j) = backward variable of state i in time t
The Baum-Welch algorithm, sometimes called the forward-backward algorithm makes use of the results derived from this algorithm to make inference. The Baum-Welch algorithm is a special form of the Expectation Maximization algorithm used for finding the maximum likelihood estimate of the parameters of the HMM. It was used in this work to train the various sized HMM parameters.
The E part of the algorithm calculates the expectation count for both the state and observation. The expectation of state count is denoted by γt(i). It is the probability of being in state at time t giving observation sequence and the model.
The expectation of transition count is denoted by εij(t). it is the probability of being in state Si at time t and state Sj at time t + 1 given an observation sequence and the model.
Based on the expectation probabilities, we can now estimate the parameters that will maximize the new model. This is the M step of the algorithm.
The new initial probability distribution can be calculated as follows.
The re-estimated transition probability distribution is also calculated as follows:
Finally, the new observation matrix is calculated using the formula below
The pseudo code of the training algorithm is represented below:
i. Begin with some randomly initialized or preselected model, π
ii. Run O through the current model to estimate the expectations of each model parameter using the forward backward algorithm.
iii. Change the model to maximize the values of each path using the new π′, α′ij and b′i(t).
iv. Repeat until change in log likelihood is less than a threshold value or when the maximum number of iterations is reached.
For global optimal results, this algorithm is usually iterated depending on the size of the dataset.
After implementing the HMM algorithms, the discretized data was parsed into the program. The dataset was trained using HMMs with cluster size from cluster 2 to 10. The likelihood of each HMM was calculated using likelihood ratio test and three clusters were identified. The likelihood ratio test of the dataset from cluster 2 to 10 is shown in Table 1 below. A positive LRT show that increasing the number of parameters is still worthwhile while a negative LRT shows that increasing the parameter would not give a better model.
The LRT becomes negative after three (3) clusters giving the optimal number as 3.
This test is used because the HMM are hierarchically nested and adding clusters will increase the likelihood of the data fitting the model until a certain point is reached when there is no significant change in the likelihood when parameters are added.
Likelihood Ratio Test is defined mathematically as:
LRT = LR – CR.
LT is the difference in the log likelihood of a more complex model to a simple model. The formula to calculate LR is illustrated below.
LR = 2(Lc – Ls)
where:
Lc = log likelihood of the complex model.
Ls = log likelihood of the simple model.
CH is the statistical chi square distribution with the number of extra parameter at a certain p –value.
This parameter is calculated by: L(Z–1).
where:
L = sequence length
Z = number of emission symbols
From the calculations below, the optimal number of clusters in the dataset based on the likelihood ratio test is 3. The log likelihoods of each cluster is also illustrated in Figure 2 below. It shows that the log likelihood increases with more parameters added initially but from cluster 3, there was no significant increase in log likelihood showing that the optimal number of clusters has been attained.
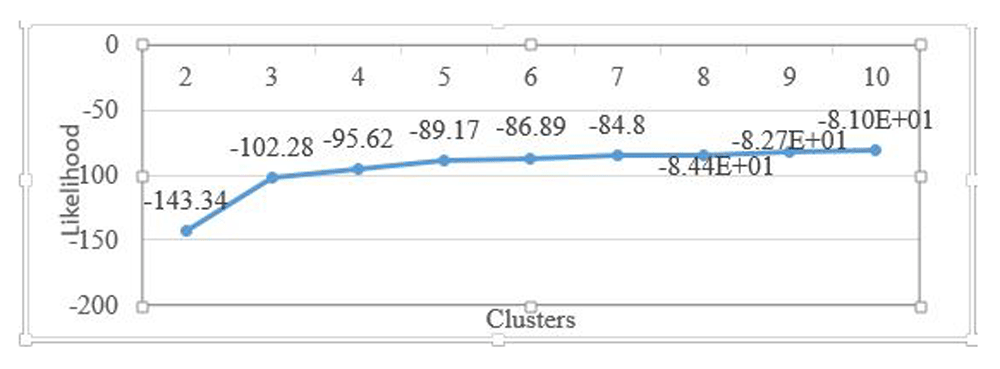
The log likelihood values increase with the number of clusters sizes. After 3 clusters, there is not a significant increase in the log likelihood.
The dataset was trained using the Baum-Welch algorithm and the probability that an observation sequence belongs to a cluster was calculated using the forward algorithm. Genes with discretized value of zeros were also removed. After the likelihood ratio test was calculated and the optimal number of clusters found, each data was then separated into clusters using the forward algorithm. The first cluster consists of 502 genes, the second cluster had 481 genes and the third cluster had 668 genes. These results are represented in the Figure 3.
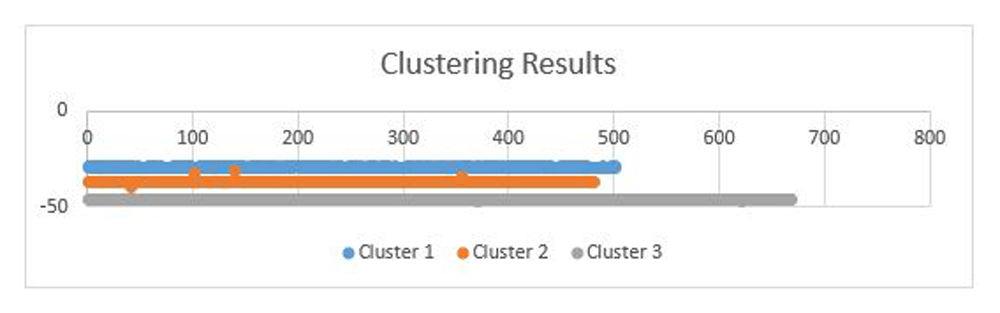
The x-axis shows the likelihood of each genes in each of the cluster and the y-axis shows the total number of genes in each cluster.
Erythrocyte membrane protein1(pfEMP1) –Clusters 1 and 2
The clustering algorithm efficiently clustered the differentially expressed genes into 3 clusters based on their functions. It is noteworthy that the most abundant genes are the erythrocyte membrane protein 1 (PfEMP1). The proteins are grouped in cluster 1 and cluster 2. PfEMP1 is an ubiquitously expressed protein during the intraerythrocytic stage of the parasite growth that determined the pathogenicity of P. Falciparum25. The virulence of P. falciparum infections is associated with the type of P. falciparum PfEMP1 expressed on the surface of infected erythrocytes to anchor these to the vascular lining. PfEMP1 represents an immunogenic and diverse group of protein family that mediate adhesion through specific binding to host receptors25. The Var genes encode the PfEMP1 family, and each parasite genome contains ~60 diverse Var genes. The differential expression of the proteins in this family has been reported to determine morbidity from P. falciparum infection26. Apart from clustering cell adhesion proteins into a sub-cluster, the algorithm also clustered the proteins involved in actin binding, transmembrane transportation and ATP binding. While the genes involved in ATP binding a largely conserved with their functions yet to be experimentally determined, the transport proteins are bet3 transport protein and aquaporin. Aquaporin is a membrane spanning transport proteins that is essential in the maintenance of fluid homeostasis and transport of water molecules and it has been identified as good therapeutic target27. Bet3 transport protein on the other hand is involved in the transport of proteins by the fusion of endoplasmic reticulum to Golgi transport vesicles with their acceptor compartment28.
The second cluster also has sub-cluster of PfEMP1 with other sub-clusters for GTP and ATP binding /ATP-dependent helicase activity as well as structural components of ribosome and ubiquitin protein ligase activity. Apart from PfEMP1, other sub-clusters are involved in protein turnover-protein synthesis and degradation29. These include the RNA helicases that prepare the RNA for translation and initiate translation, the ribosomal and GTP binding proteins that are integral part of the ribosome assembly involved in translation and the ubiquitin-conjugating enzymes are carry out the ubiquitination reaction that targets a protein for degradation via the proteasome30–32.
The genes coding for proteins involved in catabolic activities such as breakdown of proteins and hydrolysis of lipids are clustered in cluster 3. This cluster also included sub-clusters for genes involved in motor activity and DNA structural elements. The DNA structural elements include histone proteins and transcriptional initiator elements which are involved in epigenetic control of gene expression33. The ability of P. falciparum to grow and multiply both in the warm-blooded humans and cold-blooded insects is known to be under tight epigenetic regulation and it has been suggested as a good therapeutic target25.
The Functional Enrichment analysis tool was used to determine functionally enriched genes in each cluster. Each cluster was loaded separately based on their unique gene identifier. Genes are matched against the UniProt background database or by using a custom database that allows users load their own predefined Gene Ontology Term (GOTerm). Statistical cut-off of enrichment analyses in FunRich software was kept as default with a p-value <0.05. We used the custom database and loaded annotations downloaded from PlasmoDB for our functional enrichment. The result of the 3 clusters is summarized in Table 2. FunRich was only able to identify 164 of the 502 genes in cluster 1. Cluster 1 has five functional annotations, 7.3% of the genes are functionally annotated with cell adhesion molecule, receptor activity, 1.2% are annotated with acting binding, 1.2% with transporter activity and 3.7% with ATP binding as shown in Figure 4. The remaining genes has no GO functions. We can deduce that since these genes are in the same cluster, they are likely to have functions as the genes with GO annotation.
Cluster 1 has four GO annotations, cluster 2 has five and cluster 3 has seven functional annotations.
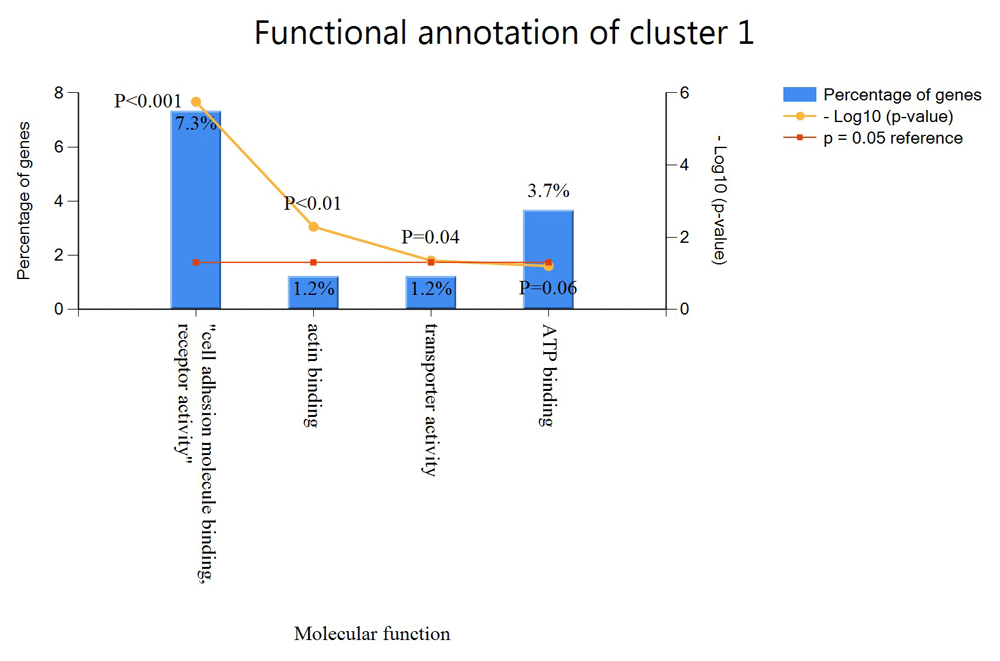
The x-axis shows the percentage genes in the cluster with specific functional annotation while the y-axis shows the function of these genes.
In cluster 2, FunRich was able to identify 308 genes. In cluster 2, 7.1% of the genes are functionally annotated with the structural constituent of the ribosome, 3.9% with cell adhesion molecule receptor activity, 1.6% with ATP binding and ATP-dependent in a helicase activity, 1.0% with ubiquitin protein ligase activity and 1.3% with GTP binding which gives a total of five functional annotations as shown in Figure 5. The rest of the genes in cluster 2 are also predicted to have similar functions as with the genes with known GO annotation.
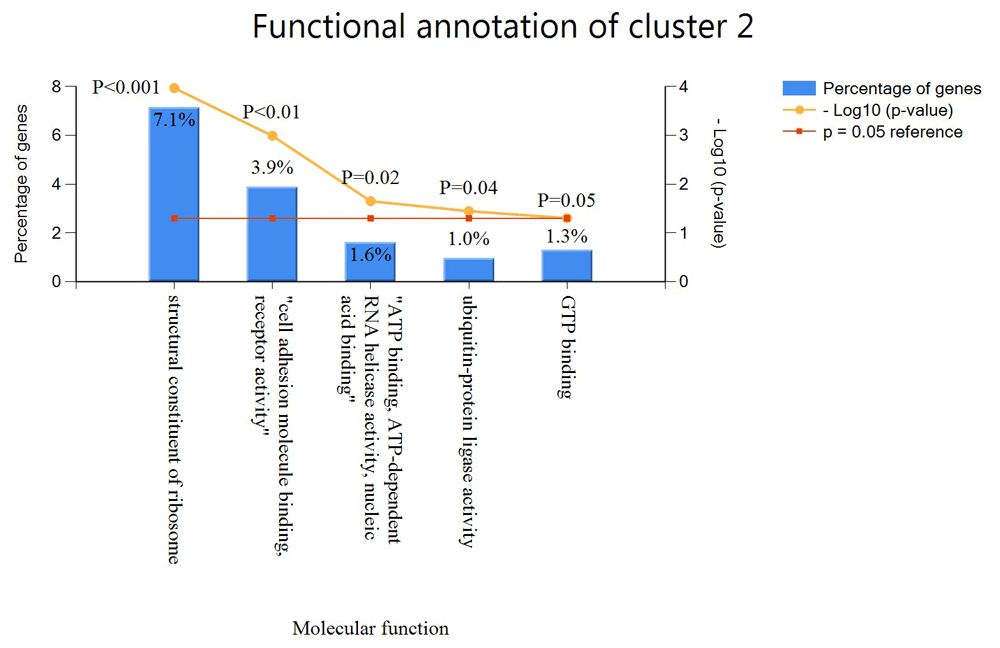
The x-axis shows the percentage genes in the cluster with specific functional annotation while the y-axis shows the function of these genes.
In cluster 3, FunRich was able to identify 249 of the 668 genes in the cluster set. This cluster has the largest GO annotation as it is enriched with seven functions. 2.8% of the genes are enriched with cysteine-type peptide activity, 2.4% with endopeptidase activity, 1.6% with hydrolase activity, 0.8% with ATP binding, action binding and monitor activity, 1.2% with DNA binding, 0.8% with unfolded protein binding and 0.8% with DNA binding, protein heterodimerization activity as shown in Figure 6. We can also deduce that in cluster 3, the genes with unknown functions are also predicted to have the same functions as with the known ones.
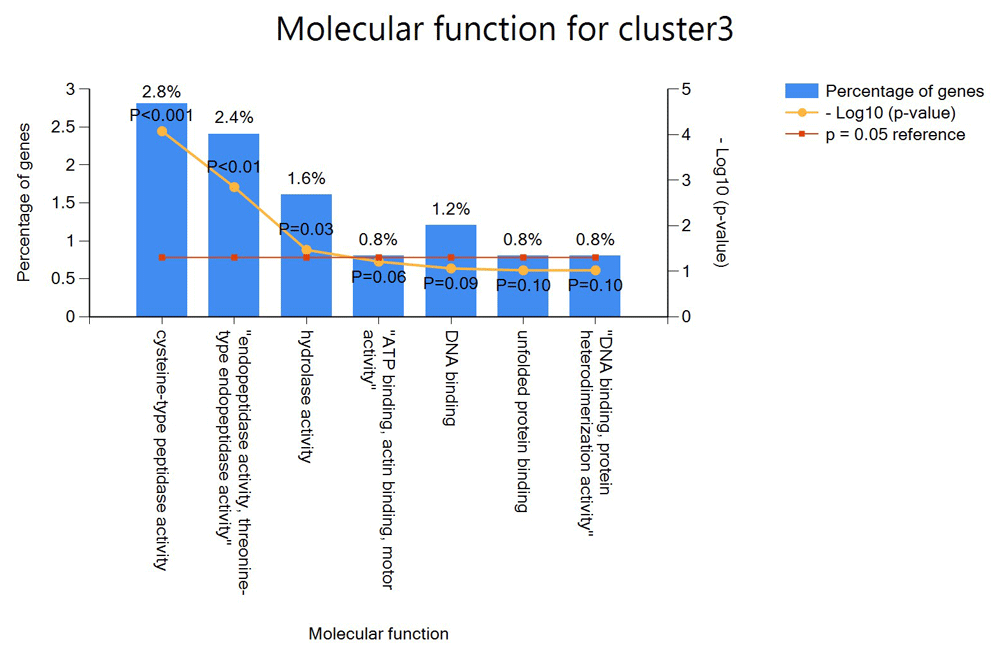
The x-axis shows the percentage genes in the cluster with specific functional annotation while the y-axis shows the function of these genes.
Clustering has been found to be a very useful technique in analyzing gene expression data. It has the ability to display large datasets in a more interpretable format. Several approaches have been developed to cluster gene expression data. The HMM has a better advantage over them because of its strong mathematical background and its ability to model gene expression data successfully. The HMM algorithms were implemented to perform cluster analysis on the P. falciparum gene expression dataset. 2000 genes were used and 3 clusters were identified. Sixteen major functional enrichment were identified for the clusters.
1. The data sets used in this study are freely available in www.ncbi.nlm.nih.gov/pubmed/20141604
Dataset 1. This .zip contains the normalized (normalized.xsl) and discretized (Discretized_Data.txt) data sets used. 10.5256/f1000research.12360.d204230
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)