Keywords
protein fragments, protein design, fragments database, structural query, triangular inequality
This article is included in the Japan Institutional Gateway gateway.
protein fragments, protein design, fragments database, structural query, triangular inequality
Thanks to the referees’ recommendations, we have improved our manuscript in this revised version. An extra paragraph was added in the introduction to describe the rationale underlying Fragger. Several citations to web servers and protein fragment databases were added to provide a broader overview of similar approaches. Some variables in the algorithm have been renamed to facilitate easy understanding. A new paragraph and one reference about choosing good reference fragments were added. Several changes were made to clarify the choice of reference fragments, and the impact of the effective cutoff values on search speed.
See the authors' detailed response to the review by Charlotte Deane and Saulo de Oliveira
See the authors' detailed response to the review by Pierre Tuffery
Nowadays, a large number of protein structures are available (122,761 as of July 2017 at RCSB) and protein fragments are frequently used in structural bioinformatics. Protein structure prediction methods such as Rosetta1, QUARK2 and EdaFold3,4 use protein fragments as building blocks. Protein fragments are also used in crystallographic phasing5–7 and model rebuilding8. The quality of protein models can be improved by combining protein fragments with molecular dynamics9. Other applications include the curation of unresolved loops in crystal structures10,11, grafting of loop sequences on protein scaffolds and other protein design algorithms12,13.
When there are too many fragments to search from, an efficient strategy is necessary to reach sub-linear search times. This problem is well-known to the chemoinformatics community, which has developed several efficient strategies to screen large databases of small molecules. For example, geometric embedding and locality sensitive hashing14, kd-trees15, a tree data structure (called µ-tree) with a heuristic16, bounds of similarity scores for chemical fingerprints17 and a proximity filter based on the logical exclusive or operator18 have all been developed to this end.
Currently, several fragment pickers19–22 and protein fragment databases23–28 are available. Of particular interest is the Super method20 that uses the lower bound of RMSD29 to screen the whole fragment space. However, our research on protein design and refinement of protein decoys for crystallographic phasing required specific options and therefore a new fragment picker.
Input: D: fragment set to query
Input: R: reference fragment set
Input: q: query fragment
Input: dq: RMSD threshold
Output: M: matching fragment set
M ← D
{fuzzy query: prune the fragment space}
for rj in R do
d ← distance(q, rj)
dinf ← d – dq
dsup ← d + dq
{distance(fi, rj) comes from the database index}
M ← {∀fi ∈ M | distance(fi, rj) ∈ [dinf, dsup]}
end for
{exact query: refine the result of pruning}
M ← {∀fi ∈ M | distance(fi, q) ≤ dq}
return M
Fragger exploits the triangular inequality of RMSD30 to prune the fragment space (Figure 1 and Algorithm 1). RMSDs are computed efficiently via the QCP method31. Fragger is written in OCaml32, except backbone RMSD computations which are performed with a new version of the C++ ranker tool from Durandal33. Computations are parallelized on multi-core computers via the Parmap library34.
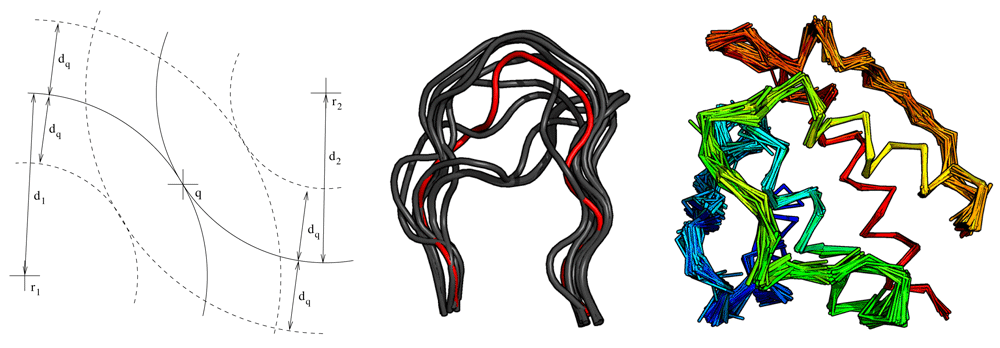
q is at distance d1 (resp. d2) from reference fragment r1 (resp. r2). Only fragments which are both within d1 ± dq of r1 and d2 ± dq of r2 will undergo an RMSD calculation. Middle: 13 residues loops that can connect residue ALA 98 to GLY 110 in chain A of PDB 1MEL. The query loop is shown in red. Only its first and last three residues were used to rank the retrieved fragments. Right: Backbone of PDB 1BKR covered with ten residue fragments from non-homologous proteins retrieved with Fragger.
Fragger allows a database to be queried with a fragment and an RMSD threshold. Matching fragments are ranked by RMSD to the query. Fragger’s ranker tool allows to compute the backbone RMSD of a single fragment versus many. Fragger can deal with residue gaps or a selection of residues from the query, create a fragment database from a set of Protein Data Bank (PDB) files, work with all fragment lengths and extract specific or randomly-chosen fragments from a database.
Compared to existing fragment pickers, some of the specific functionalities required by users include:
Outputing only the N best or N first found fragments matching a query (this can make a query terminate faster)
Constraining the amino acid sequences allowed to match a query (for loop grafting; such filtering is applied after RMSD pruning of the fragment space)
Reading and writing PDB fragments from/to a binary format (faster than reading/writing regular PDB files)
Preventing a list of PDB codes from matching a query
Automatically varying the RMSD threshold to the query until a given number of fragments is reached.
Tests were performed on one core of a 2.4GHz Intel Xeon workstation with 12GB of RAM running Ubuntu Linux 12.04. The PDB dataset is composed of all proteins determined by X-ray, without highly similar sequences (30% sequence identity cutoff) in order to create a challenging set of fragments to benchmark a protein design algorithm. It contains 13,554 PDBs. PDBs were extracted from the protein databank website using the advanced search tab and ticking the "Retrieve only representatives at 30% sequence identity" box. Querying with a three (resp. nine) residues fragment takes at least 6.75s (resp. 5.2s).
Query times vary with the query fragment, reference fragments, indexed proteins and RMSD tolerance to the query. In general, the longer the required fragment length and the smaller the RMSD tolerance, the faster the query.
Reference fragments can be chosen randomly. Pruning of the search space is better if there are at least three reference fragments, far from each other. Once a RMSD index has been computed for a randomly chosen fragment (fi), taking the furthest fragment from it (fj) and the median fragment (fk) would give three acceptable reference fragments. For interested contributors, some good heuristics can be found in the literature but were not implemented in Fragger, like Brin’s greedy algorithm35.
For one time tasks, it is not necessary to create RMSD indices and actually query a database, as fragments extraction and RMSD computations are fast enough. For example, it takes only 15s to generate all (41,200) fragments of 13 residues starting with alanine and ending with glycine (middle of Figure 1). Ranking them to the query takes 1.5s. When working on PDB files, the ranker tool included with Fragger can compute 66,580 (resp. 23,784) RMSD/s on the backbone of three (resp. nine) residue fragments. These numbers become 304,149 (resp. 138,744) RMSD/s when working on Fragger’s binary-encoded PDBs. In the future, it might be possible to improve the performance of Fragger by incorporating a faster score than RMSD, such as BCscore36.
Fragger can be useful for protein design, loop grafting and retrieval of candidates to rebuild low-confidence regions of protein models6.
All data underlying the results are available as part of the article and no additional source data are required.
Fragger can be downloaded from: https://github.com/UnixJunkie/fragger
Archived source code at the time of publication: https://zenodo.org/record/877320
Software license: LGPL.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)