Department of Physics and Astronomy, Michigan State University, East Lansing, USA
Lisa J Lapidus
Roles:
Conceptualization,
Writing – Original Draft Preparation,
Writing – Review & Editing
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
In this review, I discuss the various methods researchers use to unfold proteins in the lab in order to understand protein folding both in vitro and in vivo. The four main techniques, chemical-, heat-, pressure- and force-denaturation, produce distinctly different unfolded conformational ensembles. Recent measurements have revealed different folding kinetics from different unfolding mechanisms. Thus, comparing these distinct unfolded ensembles sheds light on the underlying free energy landscape of folding.
Corresponding author:
Lisa J Lapidus
Competing interests:
The author declares that she has no competing interests.
Grant information:
The author declares that this work was supported by the National Science Foundation’s Division of Molecular and Cellular Biosciences – 1243654.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ever since Anfinsen discovered that a protein can be reversibly folded and unfolded outside of a cell1, researchers have been investigating the folding process in vitro, confident in the knowledge that they were trying to understand a physical process of how the polypeptide chain finds a lowest free energy state. The free energy of the folded state comprises a balance of enthalpy and entropy of the protein and the surrounding solvent. The success of structural biology methods over the past few decades has focused attention on the beautiful structures of the folded states that have been determined for many sequences. In contrast, the unfolded “state”, really an ensemble of disordered conformations, was generally regarded to be highly generic, completely random, and rapidly rearranging2,3. Recent advances in nuclear magnetic resonance (NMR) and optical methods have started to change this view and detail how different sequences and different solvent conditions can alter the ensemble4–9.
Since the unfolded state was treated generically, it seemed not to matter how one obtained it, especially if the goal was to leave it as quickly as possible, as is done in kinetic folding experiments. This review will discuss the various methods that are commonly used to unfold proteins and how similar are the resulting ensembles. I will conclude with a discussion of whether the choice of unfolding method then affects the subsequent folding process.
Chemical denaturants
It remains true that chemical denaturants, guanidine hydrochloride and urea, are the most generally applicable methods of completely unfolding proteins. Both of these molecules have a low molecular weight and are extremely soluble, such that 6–8 molar concentrations will denature virtually any protein. Computational studies of polypeptides interacting with these molecules have revealed some aspects of their denaturing power10, although far more is understood about urea. The molecular dynamics of an unfolded protein indicate that urea readily forms hydrogen bonds with the peptide backbone, disrupts native contacts, and makes extended conformations favorable11. Simulations comparing urea and guanidine on the same protein find that guanidine does not make many hydrogen bonds but does disrupt hydrophobic interactions within the native state, particularly between aromatic side chains12.
A number of studies of various proteins in high denaturant have shown that these chains are acting as self-avoiding random polymers13,14. Measurement of intramolecular contact of unstructured peptides in water with guanidine and water with urea showed that a wormlike chain with excluded volume is a better model than a freely jointed chain, but the persistence length (4–6 Å) and excluded diameter (4 Å) are sufficiently small that a freely jointed chain is a good model for proteins of any reasonable length15,16. The intramolecular diffusion coefficients measured by these same experiments reveal values in the 10-6 cm2/s range for all sequences in high denaturant, about the same as the translational diffusion coefficient for objects of this size17–20. Thus, the view of unfolded proteins as completely random, freely diffusing polymers appears to be justified in high denaturant.
The difficulty with using denaturant as an unfolding mechanism is the technical challenges with rapidly diluting it to prompt refolding in a kinetic experiment. The dilution or mixing time is a period in which the solution conditions are not in equilibrium and folding kinetics are not in response to a known set of conditions. Conventional stopped-flow mixers have “dead times”, the time during which measurement is not possible, of 1–5 ms that are determined by the turbulence induced in the mixing process, yet folding may still be occurring. Smaller turbulent mixers have pushed this dead time down to as low as 30 μs21,22. Laminar flow mixers developed in my lab have eliminated turbulence and mix as fast as 2–4 μs23–26.
Temperature
It has long been known that proteins generally unfold at temperatures higher than the basal temperature of the organism in which it evolved. Therefore, the melting temperatures of proteins from thermophilic organisms are typically higher than their homologs from mesophilic organisms27–29. It has been predicted that cold denaturation, protein unfolding that occurs as the temperature is lowered30–36, is a feature of protein stability, but, practically, the lower melting temperature is rarely observable above 0°C, so water will freeze before the protein will unfold.
The appeal of using heat as a denaturant is that it is completely understood how it affects the protein on the atomic level. It is also the natural denaturant for computational studies, since heat is already accounted for in molecular dynamics simulations. In terms of the relative contributions to the change in the Gibbs free energy between the folded state and unfolded state, ΔG=ΔH-TΔS, as the temperature increases, ΔG will decrease until eventually it becomes less than zero and the unfolded state has a lower free energy than the folded state. However, the absolute free energy of the unfolded state need not remain constant with increasing temperature. In particular, the hydrophobic effect should get stronger with temperature37,38. Therefore, a protein that is unfolded at an elevated temperature may still have strong intramolecular interactions within the unfolded state that make the unfolded conformations less than completely random.
Temperature is also a natural control variable in experiments. About 20 years ago, the development of laser temperature jump (T-jump) techniques allowed the first observations of protein folding on the ns–μs time scales by adding an IR pulse of light to a protein solution to rapidly raise the temperature ~10°C in ~10 ns39–41. However, temperature may be only increased because the laser pulse adds heat. Reducing the temperature generally requires a much slower diffusive process that takes milliseconds to equilibrate. Therefore, the kinetic process that is observed is usually dominated by the unfolding process. To extract direct folding rates, researchers have usually used a two-state model of the folding process in which the relative population at each temperature is known from equilibrium measurements42–45. However, it is possible that a two-state model is not a good approximation of the folding free energy landscape46. Furthermore, given that laser T-jump is limited to increases of ~10°C and most proteins unfold at temperatures well above physiological, neither the start nor the end of the experiment is typically under strong folding conditions (i.e. 37°C).
Pressure
While few organisms undergo significant changes in pressure over their lifetime, pressure is an attractive unfolding mechanism because, like temperature, it is completely understood physically and can be simulated computationally. Under very high pressures (1–3 kbar or 100–300 MPa), voids within a protein’s folded structure become unstable, causing the protein to unfold47. The contribution to the change in free energy due to pressure is given as pΔV. The change in partial molar volume upon unfolding, ΔV, is typically negative, making the free energy of the unfolded state lower than the folded state. The kinetics of unfolding are typically extremely slow, orders of magnitude slower than unfolding with denaturant or temperature48,49. This allows the use of careful NMR measurements to map which residues change structure first and can sometimes find complex folding dynamics. Often intermediates can be detected as well as complete unfolding50. As with T-jump, it is generally difficult to rapidly depressurize to induce refolding. One technique has been demonstrated recently by Dumont et al., in which the pressure cell is irreversibly broken for each measurement but achieves depressurization within 1 μs51,52.
Force
About 20 years ago, several groups demonstrated that individual molecules could be unfolded using an atomic force microscope or optical tweezers53–55. In early measurements, proteins were immobilized on a surface and were often constructed of multiple identical domains in tandem in order to confirm the signal was real56,57. Significant improvements over the past two decades have eliminated the need for a surface (though proteins are now attached to micron-sized beads) and made detection of single-domain unfolding routine, typically using laser tweezers to trap the bead58,59. At the state of the art, these instruments can detect forces of as low as a few pN of extensions as small as ~4 Å on time scales as short as 10 μs (these limits are convolved)60–62. Like temperature and pressure, this unfolding mechanism is well understood physically, though the time-range over which these experiments are typically performed (10–100 nm/s stretching speeds) is somewhat longer than typical simulation times. The appeal of these types of measurements is two-fold: 1) they are naturally single-molecule measurements, allowing the researcher to explore heterogeneity in folding pathways62–65, and 2) the unfolded conformation eventually reached is very well defined, a completely extended polymer65. However, like many single-molecule measurements, they are typically performed at forces near the folding midpoint to allow the observation of many folding and unfolding events. If folding is induced at a low force, the instrument typically has no resolution to observe the event.
Discussion
As the descriptions above have made clear, each of these unfolding mechanisms produces an ensemble of conformations that is distinct from the others. A logical question is, should this matter? While this question does not yet have a complete answer, there are tantalizing indications that it does. For example, Lin et al. measured folding/unfolding kinetics after T-jump to the same final temperature from different initial temperatures66. For a fast two-state folder, tryptophan zipper, the kinetics were the same regardless of the starting condition, but for a protein without a significant free energy barrier between folding and unfolding, BBL, the kinetics were distinguishable. My lab, in collaboration with Bill Eaton’s lab at the NIH, demonstrated that the folding rates of the villin headpiece subdomain (HP35), one of the fastest known folders, are 5-fold slower for folding after dilution of denaturant compared to laser T-jump67. We explained this discrepancy by the Thruway Search Model first described by Ghosh and Dill68, in which the number of paths to the native state is higher and the intrachain energy of the unfolded state is lower for the chemical-denatured ensemble compared to the heat-denatured ensemble. In a study of another fast-folding protein, lambda repressor, T-jump folding rates are about 2–3-fold higher than rapid mixing folding rates69. In a different mutant of lambda, T-jump and P-jump (dropping the pressure from 1 kbar to 1 bar) yield different kinetics that were attributed to a misfolded packing of one helix accessible only when exiting out of the high-pressure unfolded state52. These discrepancies are not a universal phenomenon: T-jump and P-jump of the Fip35 fragment of a WW domain yielded very similar kinetics, as does T-jump and rapid dilution of denaturant of a different WW domain, Pin170,71.
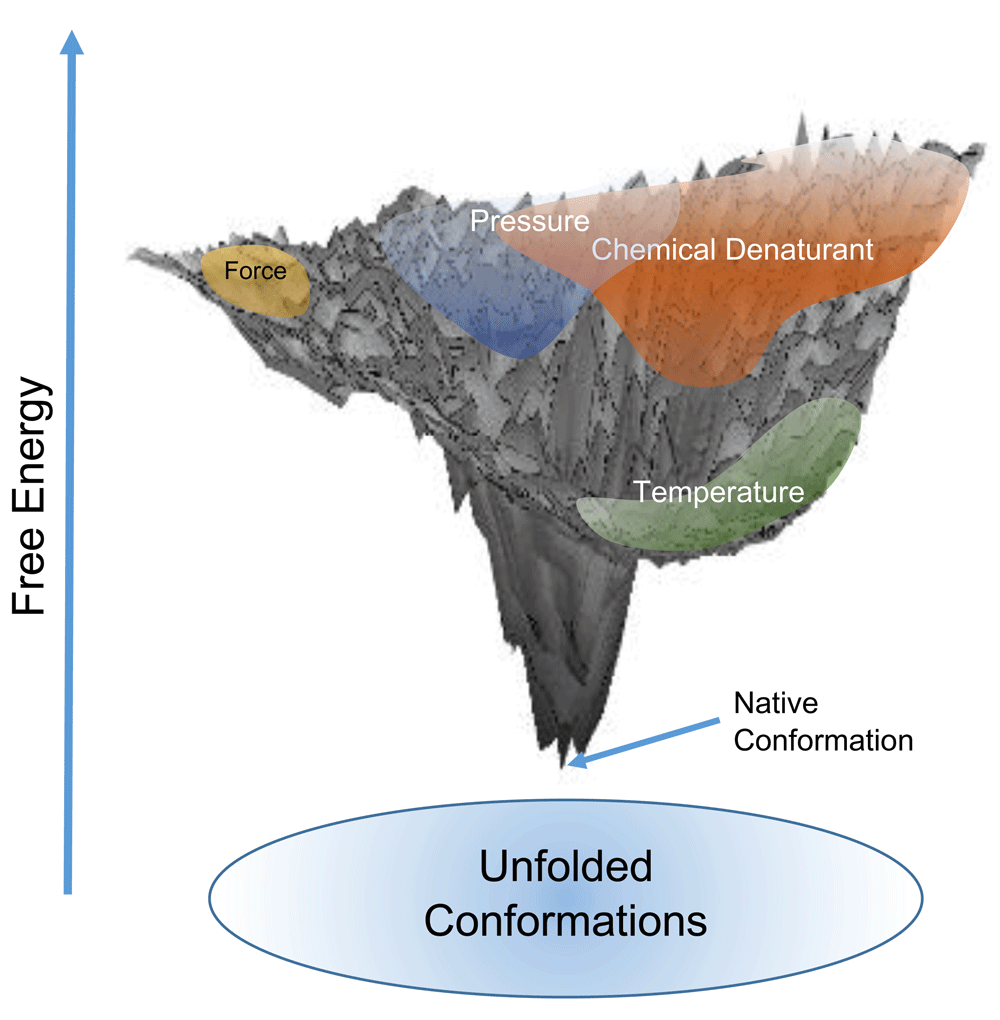
A comprehensive view of these comparisons between unfolded ensembles would suggest the complete ensemble of an unfolded protein is very broad and these various unfolding mechanisms are populating only a subset of the ensemble. Putting this in an energy landscape picture, as shown schematically in Figure 1, the top of the protein funnel is very wide and not entirely accessible by all methods. This also means that the approach to the native state from different sides of the funnel may yield different predominant pathways and folding kinetics.
Figure 1. Schematic of the free energy landscape of folding of a protein.
Unfolded ensembles by different techniques occupy distinct regions on the unfolded landscape.
Thus, from a physical perspective, the protein folding problem looks very different for any particular protein depending on how (or from where) you start the process. But from a biological perspective, should one care? The study of protein folding in vivo is still in its infancy, so we have very little idea what the unfolded ensemble looks like in a cell. Various studies have shown that thermal stability can shift up or down in crowded in vivo conditions compared to dilute in vitro conditions, depending on the sequence72–74. Danielsson and colleagues have speculated that the devil is in the details: exactly how strongly the unfolded ensemble prefers to interact with nearby proteins in a cell will determine how stability will shift75. Furthermore, it is easy to imagine that the conformational ensemble of a nascent chain emerging from the ribosome looks distinct from the ensemble achieved after unfolding within a chaperone such as GroEL/GroES. Therefore, we may expect initial folding and refolding of the same protein to use different pathways. From the perspective of biology, we should very much care how a protein is unfolded.
In conclusion, until we have a completely predictive model of protein folding, in which all folding pathways and the final folded structure are predicted by primary sequence, we have to make educated guesses of what unfolded conformations are accessible under certain conditions for a particular protein. It certainly seems sensible to continue to investigate the entire unfolded landscape under a variety of unfolding mechanisms to fully understand how protein folding depends on where you start.
Abbreviations
NMR, nuclear magnetic resonance; T-jump, temperature jump.
Competing interests
The author declares that she has no competing interests.
Grant information
The author declares that this work was supported by the National Science Foundation’s Division of Molecular and Cellular Biosciences – 1243654.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgements
The author would like to thank Matthew Comstock, Catherine Royer, and members of their research group for many helpful discussions.
2.
Smith LJ, Fiebig KM, Schwalbe H, et al.:
The concept of a random coil. Residual structure in peptides and denatured proteins.
Fold Des.
1996; 1(5): R95–106. PubMed Abstract
| Publisher Full Text
3.
Kauzmann W:
Some factors in the interpretation of protein denaturation.
Adv Protein Chem.
Elsevier; 1959; 14: 1–63. PubMed Abstract
| Publisher Full Text
4.
England JL, Haran G:
Role of solvation effects in protein denaturation: from thermodynamics to single molecules and back.
Annu Rev Phys Chem.
2011; 62: 257–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
6.
McCarney ER, Kohn JE, Plaxco KW:
Is there or isn't there? The case for (and against) residual structure in chemically denatured proteins.
Crit Rev Biochem Mol Biol.
2005; 40(4): 181–9. PubMed Abstract
| Publisher Full Text
10.
Camilloni C, Rocco AG, Eberini I, et al.:
Urea and guanidinium chloride denature protein L in different ways in molecular dynamics simulations.
Biophys J.
2008; 94(12): 4654–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
11.
Candotti M, Esteban-Martín S, Salvatella X, et al.:
Toward an atomistic description of the urea-denatured state of proteins.
Proc Natl Acad Sci U S A.
2013; 110(15): 5933–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
12.
Parui S, Manna RN, Jana B:
Destabilization of Hydrophobic Core of Chicken Villin Headpiece in Guanidinium Chloride Induced Denaturation: Hint of π-Cation Interaction.
J Phys Chem B.
2016; 120(36): 9599–607. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
13.
Kohn JE, Millett IS, Jacob J, et al.:
Random-coil behavior and the dimensions of chemically unfolded proteins.
Proc Natl Acad Sci U S A.
2004; 101(34): 12491–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
14.
Yoo TY, Meisburger SP, Hinshaw J, et al.:
Small-angle X-ray scattering and single-molecule FRET spectroscopy produce highly divergent views of the low-denaturant unfolded state.
J Mol Biol.
2012; 418(3–4): 226–36. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Buscaglia M, Lapidus LJ, Eaton WA, et al.:
Effects of denaturants on the dynamics of loop formation in polypeptides.
Biophys J.
2006; 91(1): 276–88. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Lapidus LJ, Steinbach PJ, Eaton WA, et al.:
Effects of Chain Stiffness on the Dynamics of Loop Formation in Polypeptides. Appendix: Testing a 1-Dimensional Diffusion Model for Peptide Dynamics.
J Phys Chem B.
2002; 106(44): 11628–40. Publisher Full Text
17.
Yasin UM, Sashi P, Bhuyan AK:
Expansion and internal friction in unfolded protein chain.
J Phys Chem B.
2013; 117(40): 12059–64. PubMed Abstract
| Publisher Full Text
18.
Chen Y, Parrini C, Taddei N, et al.:
Conformational properties of unfolded HypF-N.
J Phys Chem B.
2009; 113(50): 16209–13. PubMed Abstract
| Publisher Full Text
19.
Singh VR, Kopka M, Chen Y, et al.:
Dynamic similarity of the unfolded states of proteins L and G.
Biochemistry.
2007; 46(35): 10046–54. PubMed Abstract
| Publisher Full Text
20.
Buscaglia M, Schuler B, Lapidus LJ, et al.:
Kinetics of intramolecular contact formation in a denatured protein.
J Mol Biol.
2003; 332(1): 9–12. PubMed Abstract
| Publisher Full Text
21.
Bilsel O, Kayatekin C, Wallace LA, et al.:
A microchannel solution mixer for studying microsecond protein folding reactions.
Rev Sci Instrum.
2005; 76: 14302. Publisher Full Text
22.
Kathuria SV, Guo L, Graceffa R, et al.:
Minireview: structural insights into early folding events using continuous-flow time-resolved small-angle X-ray scattering.
Biopolymers.
2011; 95(8): 550–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
23.
Hertzog DE, Ivorra B, Mohammadi B, et al.:
Optimization of a microfluidic mixer for studying protein folding kinetics.
Anal Chem.
2006; 78(13): 4299–306. PubMed Abstract
| Publisher Full Text
25.
Lapidus LJ, Yao S, McGarrity KS, et al.:
Protein hydrophobic collapse and early folding steps observed in a microfluidic mixer.
Biophys J.
2007; 93(1): 218–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
26.
Yao S, Bakajin O:
Improvements in mixing time and mixing uniformity in devices designed for studies of protein folding kinetics.
Anal Chem.
2007; 79(15): 5753–9. PubMed Abstract
| Publisher Full Text
32.
Oshima H, Yoshidome T, Amano K, et al.:
A theoretical analysis on characteristics of protein structures induced by cold denaturation.
J Chem Phys.
2009; 131(20): 205102. PubMed Abstract
| Publisher Full Text
33.
Pastore A, Martin SR, Politou A, et al.:
Unbiased cold denaturation: low- and high-temperature unfolding of yeast frataxin under physiological conditions.
J Am Chem Soc.
2007; 129(17): 5374–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
35.
Privalov PL, Griko YuV, Venyaminov SYu, et al.:
Cold denaturation of myoglobin.
J Mol Biol.
1986; 190(3): 487–98. PubMed Abstract
| Publisher Full Text
36.
Yoshidome T, Kinoshita M:
Hydrophobicity at low temperatures and cold denaturation of a protein.
Phys Rev E Stat Nonlin Soft Matter Phys.
2009; 79(3 Pt 1): 30905. PubMed Abstract
| Publisher Full Text
37.
Bryngelson JD, Onuchic JN, Socci ND, et al.:
Funnels, pathways, and the energy landscape of protein folding: a synthesis.
Proteins.
1995; 21(3): 167–95. PubMed Abstract
| Publisher Full Text
39.
Thompson PA, Eaton WA, Hofrichter J:
Laser temperature jump study of the helix<==coil kinetics of an alanine peptide interpreted with a 'kinetic zipper' model.
Biochemistry.
1997; 36(30): 9200–10. PubMed Abstract
| Publisher Full Text
40.
Vitols SE, Chapin C, Callender RH, et al.:
Fast folding dynamics in peptides initiated by laser-induced temperature jumps.
Biophys J.
1997; 72(2): WP369–WP369.
41.
Ballew RM, Sabelko J, Reiner C, et al.:
A single-sweep, nanosecond time resolution laser temperature-jump apparatus.
Rev Sci Instrum.
1996; 67: 3694–9. Publisher Full Text
45.
Bunagan MR, Gao J, Kelly JW, et al.:
Probing the folding transition state structure of the villin headpiece subdomain via side chain and backbone mutagenesis.
J Am Chem Soc.
2009; 131(21): 7470–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
46.
Liu F, Du D, Fuller AA, et al.:
An experimental survey of the transition between two-state and downhill protein folding scenarios.
Proc Natl Acad Sci U S A.
2008; 105(7): 2369–74. PubMed Abstract
| Publisher Full Text
| Free Full Text
51.
Dumont C, Emilsson T, Gruebele M:
Reaching the protein folding speed limit with large, sub-microsecond pressure jumps.
Nat Methods.
2009; 6(7): 515–9. PubMed Abstract
| Publisher Full Text
52.
Prigozhin MB, Liu Y, Wirth AJ, et al.:
Misplaced helix slows down ultrafast pressure-jump protein folding.
Proc Natl Acad Sci U S A.
2013; 110(20): 8087–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
53.
Rief M, Gautel M, Oesterhelt F, et al.:
Reversible unfolding of individual titin immunoglobulin domains by AFM.
Science.
1997; 276(5315): 1109–12. PubMed Abstract
| Publisher Full Text
54.
Kellermayer MS, Smith SB, Granzier HL, et al.:
Folding-unfolding transitions in single titin molecules characterized with laser tweezers.
Science.
1997; 276(5315): 1112–6. PubMed Abstract
| Publisher Full Text
55.
Tskhovrebova L, Trinick J, Sleep JA, et al.:
Elasticity and unfolding of single molecules of the giant muscle protein titin.
Nature.
1997; 387(6630): 308–12. PubMed Abstract
| Publisher Full Text
56.
Carrion-Vazquez M, Oberhauser AF, Fowler SB, et al.:
Mechanical and chemical unfolding of a single protein: a comparison.
Proc Natl Acad Sci U S A.
1999; 96(7): 3694–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
57.
Lenne PF, Raae AJ, Altmann SM, et al.:
States and transitions during forced unfolding of a single spectrin repeat.
FEBS Lett.
2000; 476(3): 124–8. PubMed Abstract
| Publisher Full Text
62.
Žoldák G, Stigler J, Pelz B, et al.:
Ultrafast folding kinetics and cooperativity of villin headpiece in single-molecule force spectroscopy.
Proc Natl Acad Sci U S A.
2013; 110(45): 18156–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
64.
Jagannathan B, Elms PJ, Bustamante C, et al.:
Direct observation of a force-induced switch in the anisotropic mechanical unfolding pathway of a protein.
Proc Natl Acad Sci U S A.
2012; 109(44): 17820–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
65.
Hinczewski M, Gebhardt JC, Rief M, et al.:
From mechanical folding trajectories to intrinsic energy landscapes of biopolymers.
Proc Natl Acad Sci U S A.
2013; 110(12): 4500–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
66.
Lin CW, Culik RM, Gai F:
Using VIPT-jump to distinguish between different folding mechanisms: application to BBL and a Trpzip.
J Am Chem Soc.
2013; 135(20): 7668–73. PubMed Abstract
| Publisher Full Text
| Free Full Text
67.
Zhu L, Ghosh K, King M, et al.:
Evidence of multiple folding pathways for the villin headpiece subdomain.
J Phys Chem B.
2011; 115(43): 12632–7. PubMed Abstract
| Publisher Full Text
68.
Ghosh K, Ozkan SB, Dill KA:
The ultimate speed limit to protein folding is conformational searching.
J Am Chem Soc.
2007; 129(39): 11920–7. PubMed Abstract
| Publisher Full Text
69.
DeCamp SJ, Naganathan AN, Waldauer SA, et al.:
Direct observation of downhill folding of lambda-repressor in a microfluidic mixer.
Biophys J.
2009; 97(6): 1772–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
71.
Izadi D, Nguyen T, Lapidus L:
Complete Procedure for Fabrication of a Fused Silica Ultrarapid Microfluidic Mixer Used in Biophysical Measurements.
Micromachines.
2017; 8(1): 16. Publisher Full Text
72.
Wang Y, Sarkar M, Smith AE, et al.:
Macromolecular crowding and protein stability.
J Am Chem Soc.
2012; 134(40): 16614–8. PubMed Abstract
| Publisher Full Text
The author declares that this work was supported by the National Science Foundation’s Division of Molecular and Cellular Biosciences – 1243654.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Lapidus LJ. Protein unfolding mechanisms and their effects on folding experiments [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):1723 (https://doi.org/10.12688/f1000research.12070.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Park C. Reviewer Report For: Protein unfolding mechanisms and their effects on folding experiments [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):1723 (https://doi.org/10.5256/f1000research.13059.r26044)
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Park C. Reviewer Report For: Protein unfolding mechanisms and their effects on folding experiments [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):1723 (https://doi.org/10.5256/f1000research.13059.r26044)
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)