Keywords
herpes virus, envelopment, egress pathway, endoplasmic reticulum, Golgi complex, intraluminal transport, brefeldin A,
herpes virus, envelopment, egress pathway, endoplasmic reticulum, Golgi complex, intraluminal transport, brefeldin A,
The Golgi complex plays a crucial role in the secretory pathway. Cargo is transported from the endoplasmic reticulum (ER) to the Golgi complex via vesicles that derive from ER exit sites (Bonifacino & Glick, 2004). Tubules are involved in anterograde as well as in retrograde transport (Lippincott-Schwartz et al., 1990; Lippincott-Schwartz et al., 1989). The sole paradigm of vesicular transport between ER and Golgi may evolve to account for the results of new technologies (Lippincott-Schwartz, 2011). Indeed, cargo may also be transported through an ER-Golgi intermediate compartment (ERGIC) (Hauri & Schweizer, 1992; Klumperman, 2000; Saraste et al., 2009). Whether the ERGIC is a stable structure, is under debate (Ben-Tekaya et al., 2005). There are also transitional elements connecting Golgi membranes to ER membranes (Pavelka & Roth, 2015; Polishchuk & Mironov, 2004; Vivero-Salmeron et al., 2008) possibly enabling direct transportation of cargo from ER cisternae into Golgi cisternae. Once in the Golgi cisternae, cargo is packaged into secretory granules for exocytotic release (Palade, 1975). Packaging of cargo is accompanied by loss of Golgi membranes. To maintain Golgi structure and function, multiple recycling processes take place (Orci et al., 1981). Although the structure and function of the Golgi complex has been investigated for decades, many uncertainties remain e.g. Golgi maturation and functionality of the trans Golgi network (TGN) (Emr et al., 2009).
The Golgi complex plays also a crucial role in herpes virus morphogenesis and intracellular transport (Roizman et al., 2014). Herpes viruses comprise the capsid, tegument and envelope, with embedded glycoproteins. Capsids are assembled in nuclei of host cells and transported to the Golgi complex concomitantly acquiring the envelope and tegument. Three diverse pathways have been proposed. In pathways 1 and 3 (summarized in Figure 11), capsids directed to the nuclear periphery are released into the perinuclear space (PNS) by budding at the inner nuclear membrane (INM) acquiring an electron dense envelope and tegument. In pathway 1, these perinuclear virions are intraluminally transported (Gilbert et al., 1994; Granzow et al., 1997; Radsak et al., 1996; Schwartz & Roizman, 1969; Stannard et al., 1996; Sutter et al., 2012; Whealy et al., 1991; Wild et al., 2002) into ER cisternae, whose membranes are connected to the outer nuclear membrane (ONM). These virions were suggested to be transported via ER-Golgi transitions into Golgi cisternae (Leuzinger et al., 2005; Wild et al., 2015; Wild et al., 2002). Importantly, intraluminal transportation requires mechanisms for preventing the viral envelope from fusion with the membrane the virions are transported along.
In pathway 2, capsids are released from the nuclear periphery into the cytoplasmic matrix via impaired nuclear pores (Leuzinger et al., 2005; Wild et al., 2005; Wild et al., 2009) or disrupted nuclear membranes (Borchers & Oezel, 1993; Klupp et al., 2011). Notably, pore impairment is the initial step in breakdown of the nuclear envelope (Terasaki et al., 2001) that takes place when HSV-1 infection proceeds (Maric et al., 2014). The capsids in the cytoplasmic matrix are transported to any site of the Golgi complex (Wild et al., 2002) and are enveloped either by wrapping (see below) or by budding into Golgi cisternae and/or vacuoles, which may enlarge to engulf multiple virions (Homman-Loudiyi et al., 2003; Leuzinger et al., 2005; Stannard et al., 1996; Sutter et al., 2012; Wild et al., 2015; Wild et al., 2002). Capsids bud, though less frequently, also at the ONM and RER membranes (Leuzinger et al., 2005; Wild et al., 2005), and are intraluminally transported as in pathway 1.
According to pathway 3, virions in the PNS are de-enveloped by fusion of the viral envelope with the ONM or with adjacent ER membranes releasing capsid and tegument into the cytoplasmic matrix. These capsids then are re-enveloped by budding at membranes of the TGN (Mettenleiter et al., 2013) and endosomes (Albecka et al., 2016; Hollinshead et al., 2012) acquiring again an envelope and tegument. Concomitantly, a small concentric transport vacuole is formed enclosing the enveloped virion. This process is referred to as wrapping (Roizman et al., 2014).
Although the phenotypes of the capsid transport across the ONM exhibit all characteristics of budding (Bonifacino & Glick, 2004; Harrison, 2015; Jahn et al., 2003; Kanaseki et al., 1997; Lee, 2010; Mayer, 2002) the de-envelopment theory is still favored. Fusion of the viral envelope with the ONM requires the glycoproteins gB and gH (Farnsworth et al., 2007). Nonetheless, various stages of capsid transportation across the ONM and ER membranes in the absence of the glycoproteins gB/gH have been shown. Furthermore, a substantial proportion of virions lacking gB/gH were reported to be in the cytoplasm and extracellular space. These two facts strongly suggest another pathway of virus particles out of the PNS. The lack of profound evidence for capsid transport across the ONM via fusion, and the growing evidence of Golgi to ER transitions, prompted us to investigate the ER and Golgi compartments by cryo-field emission scanning electron microscopy (Cryo-FESEM) and transmission electron microscopy (TEM), employing protocols for improved spatial and temporal resolution (Ellinger et al., 2010; Mueller, 1992). We show that the Golgi complex is a tightly packed organelle, that ER to Golgi transitions are formed, and that virions aggregate within the ER after exposure cells to brefeldin A (BFA), which disintegrates the Golgi complex within minutes (Hess et al., 2000), suggesting an intraluminal transportation route. Moreover, we observe that intraluminal virions are densely coated with a proteinaceous layer that arises during budding. Therefore, we propose that the significance of the dense coat is protecting the viral envelope from fusion with the membranes along which virions are transported, in a similar manner as clathrin protects coated vesicles from fusion.
Vero cells and MDBK cells (European Collection of Cell Cultures) were grown in Dulbecco’s modified minimal essential medium (DMEM; Gibco, Bethesda, MD, USA) supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml) and 10% fetal bovine serum (FBS; Gibco). The Us3 deletion mutant R7041(ΔUs3) and the repair mutant R2641 (Longnecker & Roizman, 1987; Purves et al., 1987) were kindly provided by Bernard Roizman (The Marjorie B. Kovler Viral Oncology Laboratories, University of Chicago, Illinois, USA). Wild-type herpes simplex virus 1 (wt HSV-1) strain F (Ejercito et al., J. Gen. Virol. 2:357–364, 1968), R7041(ΔUs3) and R2641 were propagated in Vero cells, and bovine herpes virus 1 (BoHV-1: Metzler et al., Arch. Virol. 87: 205–217, 1986) in MDBK cells. Virus yields were determined by plaque titration.
50 μm thick sapphire disks (Bruegger, Minusio, Switzerland) measuring 3 mm in diameter were coated with 8–10 nm carbon, obtained by evaporation under high vacuum conditions to enhance cell growth and to facilitate detachment of cells from the sapphire disks after embedding. Vero and MDBK cells were grown for 2 days on sapphire disks placed in 6 well plates. Cells were inoculated with R7041(ΔUs3), the repair mutant R2641, wt HSV-1 or BoHV-1 at a MOI of 5, incubated at 37°C, and fixed at 8 to 20 hpi by adding 0.25% glutaraldehyde to the medium prior to freezing in a high-pressure freezing unit (HPM010; Science Services, Munich, Germany) and processed as described in detail (Wild, 2008). In brief, the frozen water was substituted with acetone in a freeze-substitution unit (FS 7500; Boeckeler Instruments, Tucson, AZ, USA) at -88°C with acetone and subsequently fixed with 0.25% glutaraldehyde and 0.5% osmium tetroxide raising the temperature gradually to +2°C to achieve good contrast of membranes (Wild et al., 2001) , and embedded in epon at 4°C followed by polymerization at 60°C for 2.5 days. After removal of sapphire disks by immersion in liquid nitrogen, serial sections of 60 to 90 nm thickness were analyzed in a transmission electron microscope (CM12; FEI, Eindhoven, The Netherlands) equipped with a CCD camera (Ultrascan 1000; Gatan, Pleasanton, CA, USA) at an acceleration voltage of 100 kV.
Cells grown on sapphire disks were inoculated with wt HSV-1 at a MOI of 5 and incubated at 37°C. Stock solution (5 mg BFA solved in 0.5 ml methanol) was diluted with medium 1:10. One µl/ml medium (1µg/ml) of this solution was added to cell cultures at 5, 8, 12 and 16 hpi. Cells were high-pressure frozen at indicated times, and prepared for TEM. To quantify virus phenotypes and their location, 10 images of cellular profiles were taken at random from ultrathin sections of monolayers exposed to BFA from 5, 8, 12 or 16 hpi to 20 hpi of 5 independent experiments. Capsids budding at the INM, ONM, ER and Golgi membranes, capsids undergoing wrapping, as wells as virions in the PNS, ER, Golgi cisternae and vacuoles were counted. The means expressed per cellular profile were compared applying a Student’s t-test using GraphPad Prism 3 software.
For determination of infectious progeny virus produced after BFA exposure, cells were grown in 10 ml Falcon flasks, inoculated with wt HSV-1 at a MOI of 5, and exposed to BFA from 5, 8, 12 or 16 h to 20 hpi. Cells were harvested at 5, 8, 12, 16 and 20 hpi for determination of infectious progeny virus by plaque titration. Since the Golgi complex reacts immediately to BFA by disintegration, infectious viruses produced after BFA exposure were considered to have derived by budding at membranes other than those of the Golgi complex.
Vero cells were grown in 25 cm2 cell culture flasks for 2 days prior to inoculation with wt HSV-1, R7041(ΔUs3) or R2641 at MOI of 5. Cells were harvested at 9 to 12 hpi by trypsinization followed by centrifugation at 150 x g for 8 min. Pellet were re-suspended in 1 ml fresh medium, collected in Eppendorf tubes and fixed by adding 0.25% glutaraldehyde to the medium. The suspension was kept in the tubes at 4°C until cells were sedimented. After removal of the supernatant, cells were frozen in a high-pressure freezing machine EM HPM100 (Leica Microsystems, Vienna, Austria) as described in detail previously (Wild et al., 2012; Wild et al., 2009). Cells were fractured at -120°C in a freeze-fracturing device BAF 060 (Leica Microsystems) in a vacuum of 10-7 mbar. The fractured surfaces were partially freeze-dried (“etched”) at -105°C for 2 min, and coated with 2.5 nm platinum/carbon by electron beam evaporation at an angle of 45°. Some specimens were coated additionally with 4 nm of carbon to reduce electron beam damage during imaging at high magnifications. Specimens were imaged in an Auriga 40 Cross Beam system (Zeiss, Oberkochen, Germany) equipped with a cryo-stage (Leica Microsystems) at -115°C and an acceleration voltage of 5 kV using the inlens secondary electron detector.
Cells were grown for 2 days on 0.17 mm thick cover slips of 12 mm in diameter (Assistent, Sondheim, Germany) and inoculated with R7041(ΔUs3), wt HSV-1 or R2641 at a MOI of 5 and incubated at 37°C. After fixation with 2% formaldehyde for 25 min at room temperature, cells were briefly washed with PBS and stored in PBS at 4°C until further processing. Then, cells were permeabilized with 0.1% Triton-X-100 at room temperature for 7 min and blocked with 3% bovine serum albumin in PBS containing 0.05% Tween 20 (PBST). To ascertain infectivity, cells were labeled with antibodies against ICP4 (Life Technologies, Carlsbad, CA; USA), and Alexa 594 (Molecular Probes, Eugene, OR, USA) as secondary antibodies. To identify the Golgi complex, cells were incubated with recombinant monoclonal antibodies against the cis-Golgi protein GM130, abcam EP892Y (Abcam, Cambridge, UK) at a dilution of 1:1000 for 2 h at room temperature followed by incubation with Alexa 488 (Molecular Probes) as secondary antibodies, diluted 1:500, for 1 h at room temperature. After staining nuclei with 4',6-Diamidino-2-phenylindol (DAPI; Roche, Mannheim, Germany), cells were embedded in glycergel mounting media (Dako North America, Carpinteria, CA, USA) and 25 mg/ml 1,4-diazabicyclo [2.2.2] octane (DABCO; Fluka, Buchs, Switzerland). Specimens were analyzed using a confocal laser scanning microscope (SP2; Leica, Wetzlar, Germany). Images were deconvolved employing the deconvolution algorithm of the program suite Huygens Essential (SVI, Hilversum, The Netherlands).
To localize the Golgi complex, we first imaged the Golgi complex by confocal microscopy after labeling the cis-face with antibodies against the Golgi protein GM 130 in Vero cells. The Golgi complex was always found close to the nucleus in HSV-1, R7041(ΔUs3), or mock infected cell (Figure 1). Next, high resolution cryo-electron microscopy of freeze fractured cells demonstrated the bell-shaped form of the Golgi complex, and its localization close to the nucleus (Figure 2). This image also shows that the Golgi complex is a complex tightly packed entity with a diameter of approximately 6 µm. The Golgi complex is separated from the cytoplasmic matrix by an intact membrane covering the whole visible surface of the cis-face. The large dimension makes clear that the detected ultrastructural details depend on a large scale on how and where the Golgi complex is hit in a given section plane for studying by TEM. Both freeze-fracture planes and thin sections of central regions show that the membrane of the outermost cisterna covers the cis-side (Figure 3).
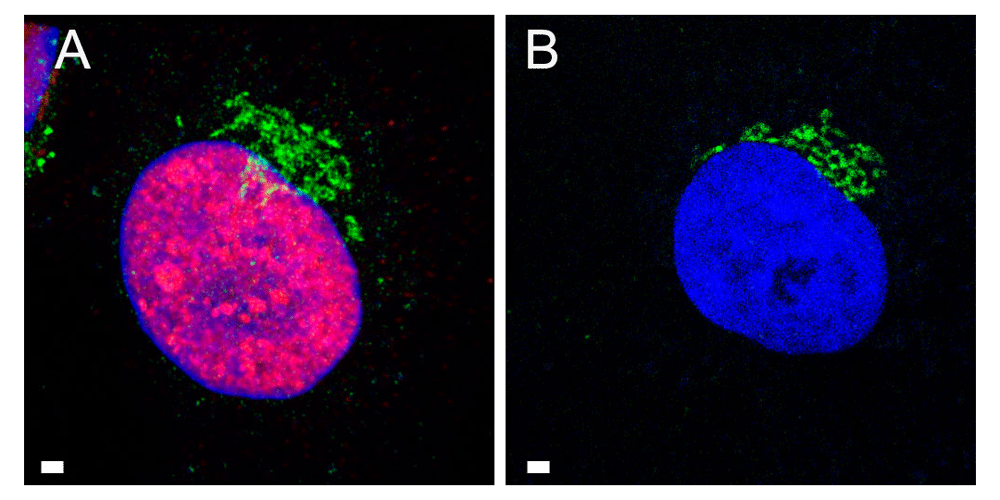
Confocal microscopy of Vero cells, immunolabeled with antibodies against the Golgi protein GM130 (green) and ICP4 (red) at 12 hpi with HSV-1 (A) and after mock infection (B), showing the Golgi complex always in a juxtanuclear position. Bars: 1 µm.
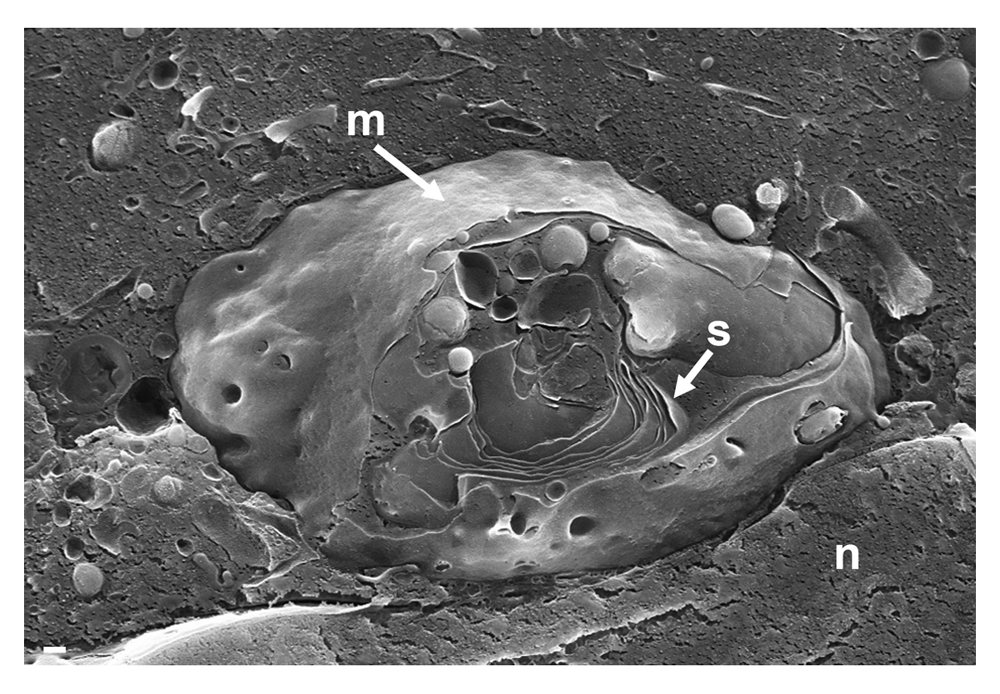
The entire visible surface is covered by an intact membrane (m) except at the part it is broken away giving view to Golgi stacks (s). Bars: 200 nm.
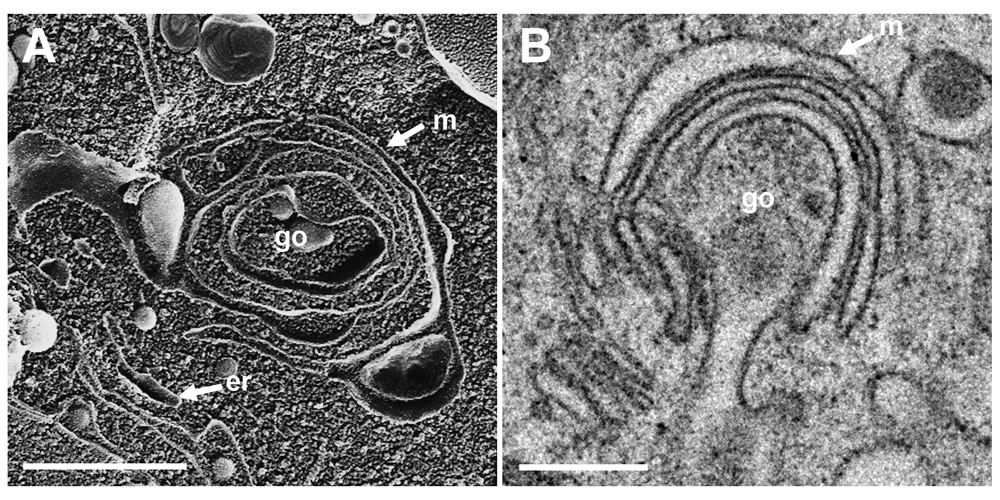
The Golgi complex, as revealed in freeze-fracture planes (A) and in thin sections (B), is entirely covered at the cis-face by the membrane (m) of the outermost cisterna. Bars: 100 nm.
The Golgi complex undergoes dramatic changes during HSV-1 infection finally resulting in fragmentation and dispersion (Campadelli et al., 1993). However, the Golgi complex is not, or only minimal, involved in envelopment of R7041(ΔUs3) capsids (Wild et al., 2015). To identify ER-to-Golgi transitions, we thus imaged the Golgi complex in serial sections through wt HSV-1 or R7041(ΔUs3) infected cells by transmission electron microscopy after rapidly freezing and freeze-substitution applying a protocol especially suitable to visualize membranes (Wild et al., 2001). A series of images show that ER membranes continue into Golgi membranes. The ER runs from the perinuclear region into the membranes of the outermost Golgi stack (Figure 4A). The very same membranes continue again into ER membranes so that this Golgi cisterna is interconnected between ER lamellae. The membranes of the adjacent stack also turn into ER membranes. ER membranes also pass somewhere into central regions of Golgi fields where they are devoid of ribosomes (Figure 4BC). ER membranes may even connect two Golgi fields (Figure 4D). The ER forms, as its name implies, a network (Figure 4C) that connects to the PNS (Figure 5A).
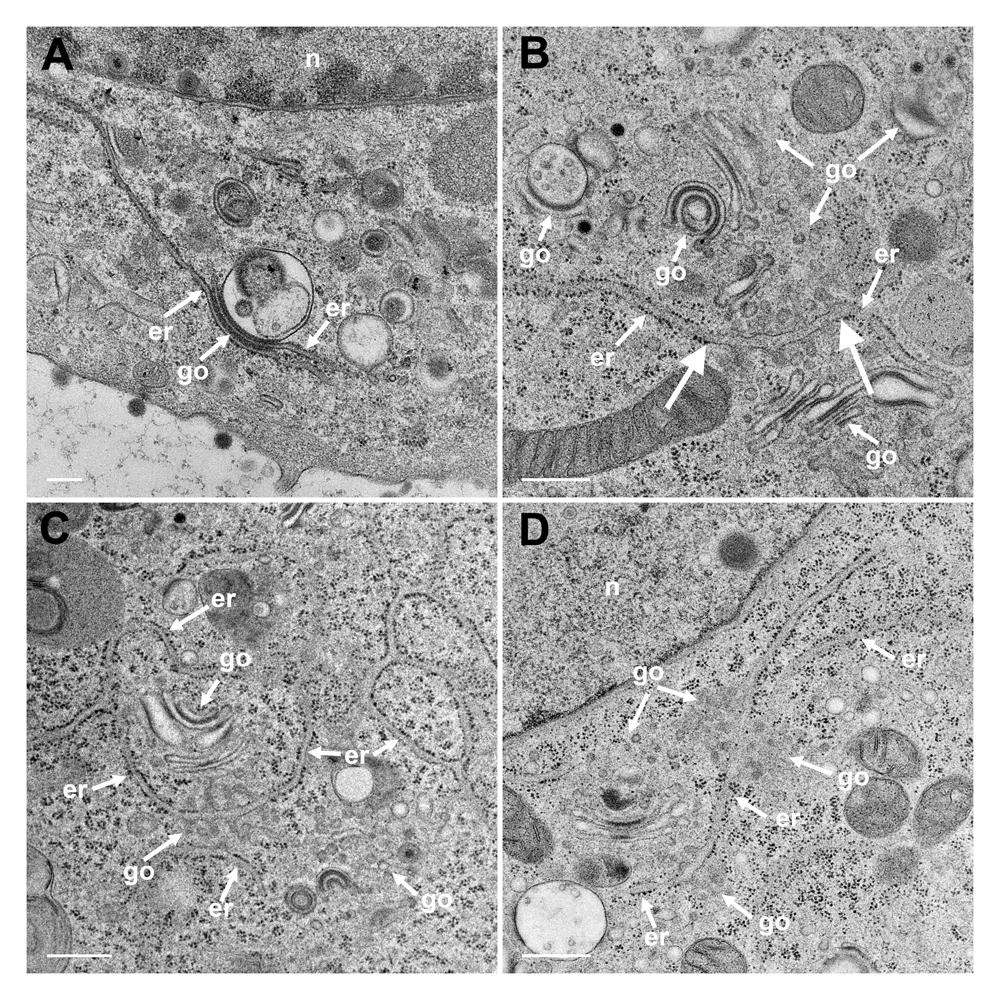
(A) The ER (er) runs from the nuclear (n) periphery towards a Golgi field (go), continuing into the membrane of the outermost stack and further into the cytoplasmic matrix. The membranes of the second stack continue also into ER membranes. (B) An ER cisterna runs through multiple small Golgi fields, whereby the ER membranes turn into Golgi membranes (thick arrows). (C) ER membranes forming a network continue into Golgi membranes. (D) ER membranes run through two Golgi fields, turning each time into Golgi membranes. Bars: 500 nm.
Virions within the ER have been repeatedly shown, the first report dating back to the late 1960ties (Schwartz & Roizman, 1969). Virions were in the PNS or anywhere in ER cisternae after HSV-1 infection (Figure 5BC) as well as in Golgi cisternae of which membranes transit into ER membranes (Figure 5B). Virions accumulate in the PNS-ER compartment late in infection (Leuzinger et al., 2005; Wild et al., 2015), in the absence of Us3 (Reynolds et al., 2002; Wisner et al., 2009) or gB/gH (Farnsworth et al., 2007) or after disintegration of the Golgi complex by BFA (Chatterjee & Sarkar, 1992; Cheung et al., 1991; Jensen & Norrild, 2002; Whealy et al., 1991). To investigate the effect of BFA on virus release out of the PNS we exposed cells to BFA at 5, 8, 12 and 16 hpi with wt HSV-1, and harvested cells at 20 hpi for quantitative electron microscopic analysis and determination of infectious progeny virus. Electron microscopy revealed dilation of the PNS and ER containing many virions and amorphous material at 15 hpi (Figure 5D). At 17 hpi, the ER was congested with virions (Figure 5E). Quantitative analysis of phenotype distribution revealed that virions accumulate in the PNS-ER compartment after BFA administration in a time dependent manner. The number of intraluminal virions was 4 times higher when BFA was added at 8 hpi but 12 times higher than it was added at 16 hpi compared to that in untreated cells (Figure 6). The number of virus particles interacting with the ONM and ER membranes in BFA exposed cells was about twice as high as in controls whereas the number of capsids in the cytoplasmic matrix did not differ significantly. We thus conclude i) that the interactions at the ONM and ER membranes are budding capsids contributing to accumulation of virions in the PNS and ER, ii) that more capsids bud at the ONM and ER membranes because no Golgi membranes are available after Golgi disintegration induced by BFA, iii) that inhibition of virion release out of the PNS-ER compartment is due to a blockage of the intraluminal transportation pathway after Golgi disintegration, and iv) that the Golgi complex delivered components to budding sites prior to its disintegration by BFA.
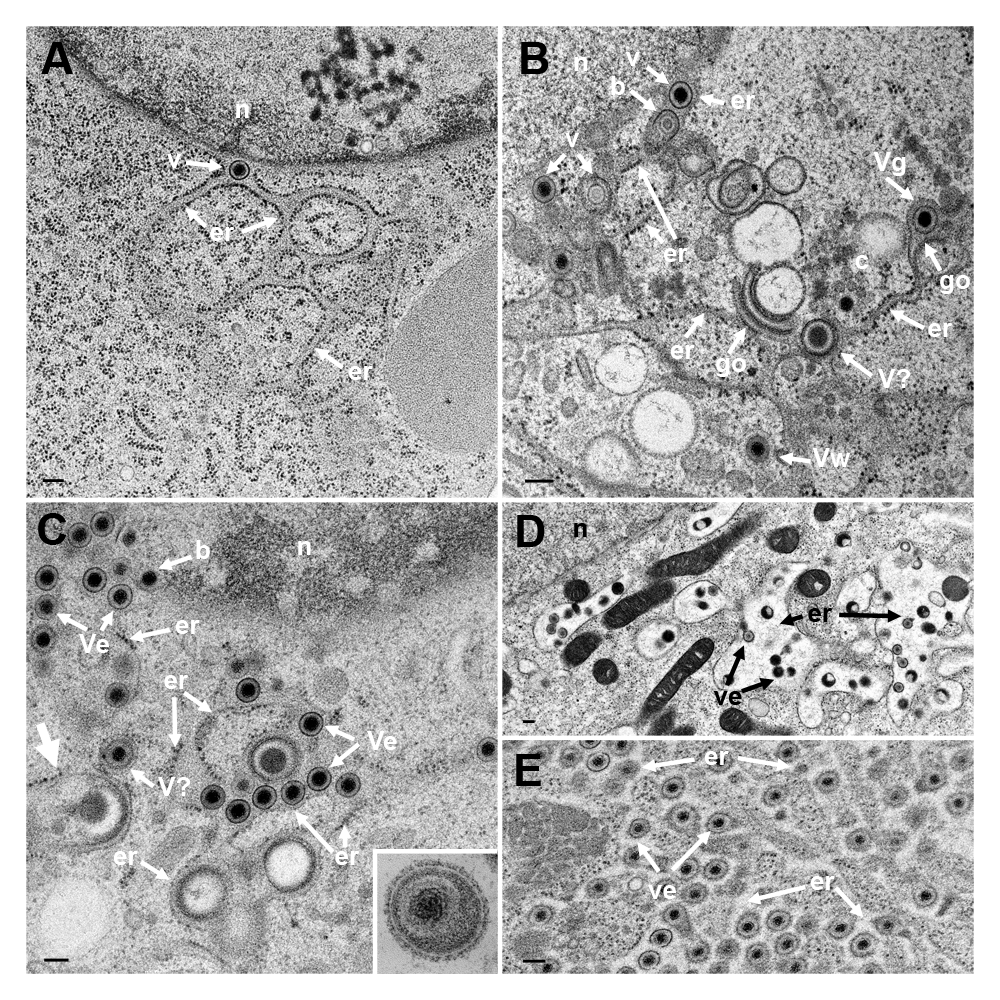
TEM of Vero cells at 9 hpi with R7041(ΔUs3) (A), at 16 hpi (B) and at 20 hpi (C) with wt HSV-1, and at 15 or 17 hpi with wt HSV-1 and BFA exposure (D and E). (A) The ER (er) runs from the nucleus (n) towards the cell periphery forming an entity with the PNS that contains a virion (v). (B) The ER contains virions. One capsid is in the stage of budding (b) into the ER. The ER continues into Golgi (go) membranes at two sites. One Golgi cisterna contains a virion (Vg), one virion has been derived by wrapping (Vw). Close to Golgi stacks, there is probably a virion (V?) of abnormal size. (C) One capsid buds (b) at the nuclear (n) periphery. The ER is dilated and filled with virions (Ve) and dense material: An ER membrane turns into a Golgi membrane (thick arrow). (D) After exposure to BFA from 8 to 15 hpi with wt HSV-1, the ER was dilated and contained some virions. (E) The ER was almost filled with virions after exposure to BFA from 8 to 17 hpi with wt HSV-1. Note that virions in the PNS and ER are covered by a dense coat hiding spikes whereas spikes are clearly apparent on virions in the extracellular space (C inset). Bars: 200 nm.
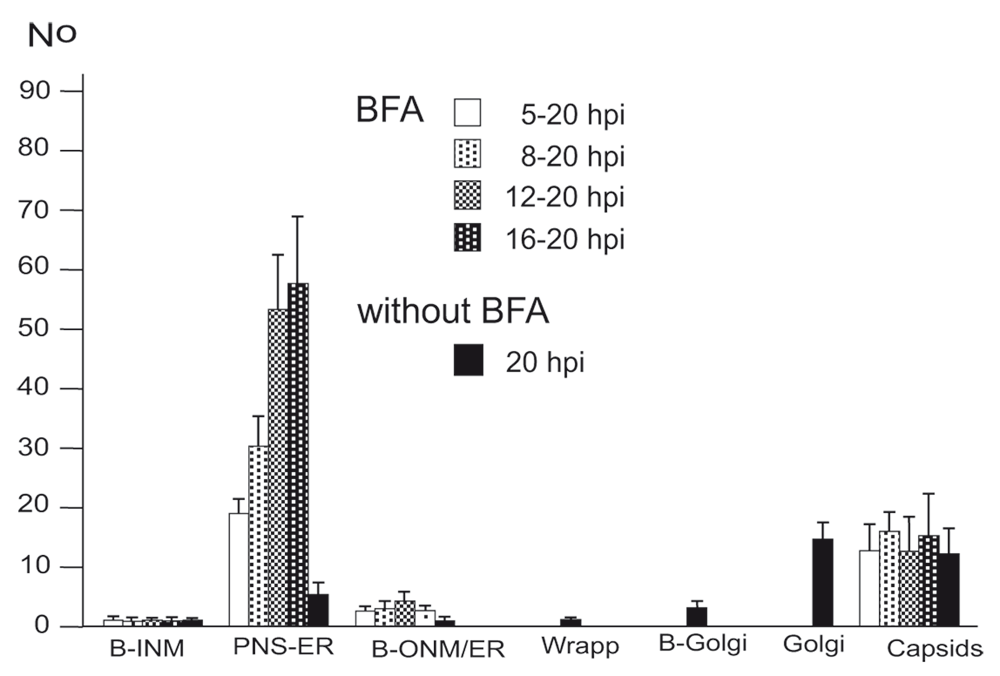
BFA was added to monolayers at 5, 8, 12 or 16 hpi (MOI of 5) and incubated until 20 hpi. For control, inoculated cells were incubated for 20 h without addition of BFA. Cells were rapidly frozen at 20 hpi and processed for electron microscopy. The phenotypes of envelopment were counted in 10 cellular profiles of 5 independent experiments: Capsids budding at the INM (B-INM), at the ONM and ER membranes (B-ONM/ER) and at the Golgi complex (B-Golgi); virions in the PNS-ER compartment (PNS-ER); virions derived by wrapping (Wrapp); virions in Golgi cisternae or large vacuoles (Golgi); capsids in the cytoplasmic matrix (capsids).
Us3 is not essential (Poon et al., 2006; Reynolds et al., 2002; Ryckman & Roller, 2004; Wisner et al., 2009). Us3 deletion mutants accumulating in the PNS are infective (Wild et al., 2015). Hence, it is reasonable to assume that wt HSV-1 virions in the PNS-ER compartment are also infective. To prove this idea, we determined infectious progeny virus by plaque titration at the time point of BFA administration and at 20 hpi. The Golgi complex completely disintegrates within less than 5 minutes after exposure of cells to BFA (Hess et al., 2000). Despite of Golgi disintegration infectious progeny virus was produced in a time depended manner. The later BFA was added the more infectious viruses were produced by 20 hpi (Figure 7). Since virions accumulated exceptionalness in the PNS-ER compartment in BFA exposed cells we conclude that virions derived by budding at nuclear membranes are infective.
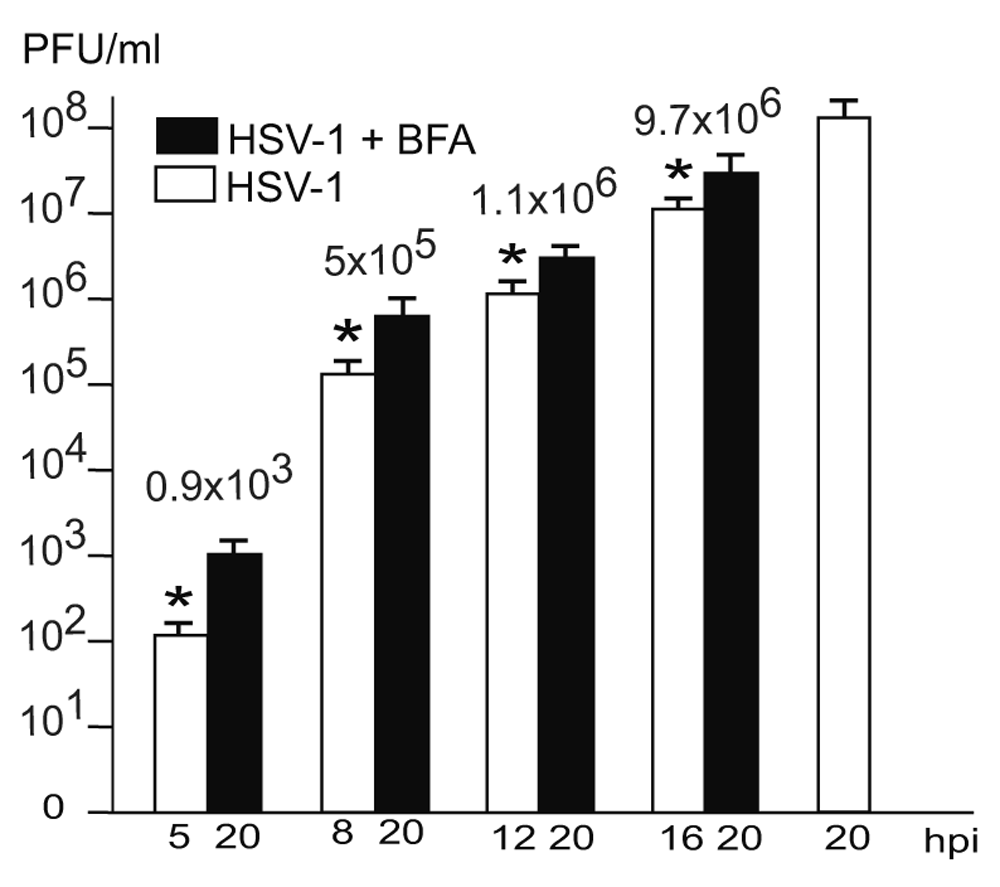
The difference between virus yields (indicated with numbers) at the time of BFA addition and harvesting at 20 hpi is considered to be due to virus production after Golgi disintegration. These infectious virions correspond to the virions accumulating in the PNS-ER compartment. n = 4, p<0.01.
The nuclear envelope is part of the ER. The outer nuclear membrane (ONM) is stubbed with ribosomes. The ONM continues into ER cisternae, which, in turn, merge with Golgi cisternae. Golgi cisternae were also found to connect to the PNS via short ER-Golgi intermediates in R7041(ΔUs3) infected Vero cells (Figure 8ABC) and BoHV-1 infected MDBK cells (Figure 8D). ER-Golgi intermediates contained virus like particles (Figure 8B). The continuum between PNS and Golgi cisternae is considered likely to serve as a direct, short and efficient pathway to transport virions form the site of budding to Golgi cisternae for packaging.
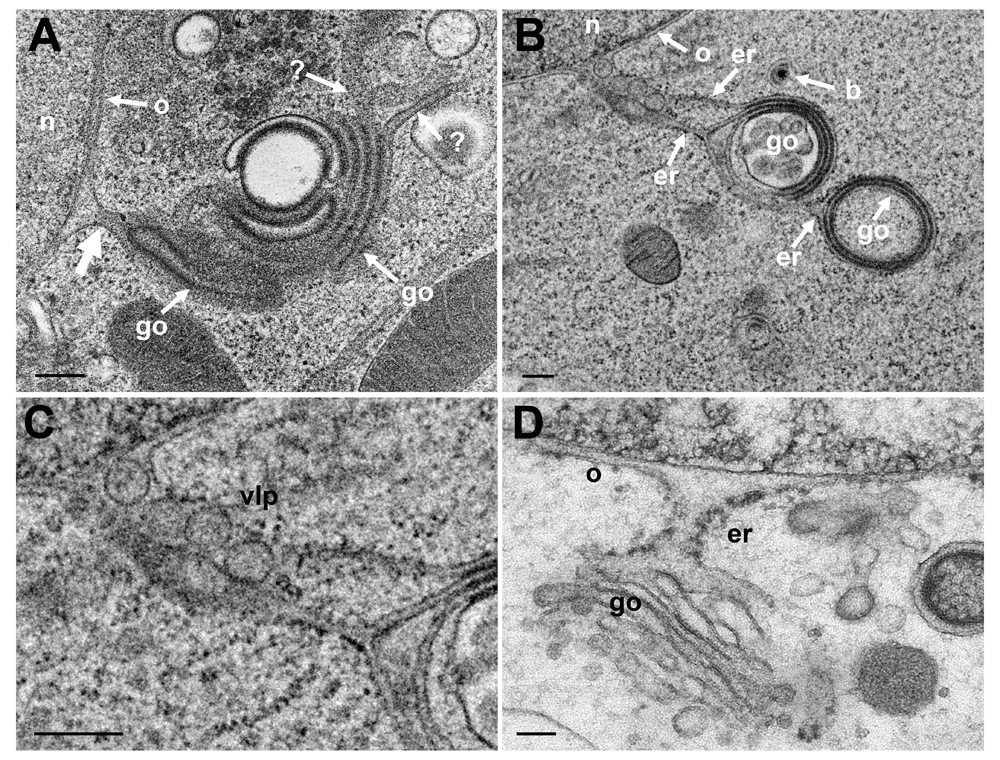
(A) Golgi (go) membranes continue (thick arrow) into the ONM (o) as well as towards the cytoplasm indicated by (?) because the destination is unknown. (B) Golgi membranes continue via ER membranes (er) into the ONM. The ER contains 4 virus-like particles. (C) Details of panel B. (D) PNS, ER and Golgi complex form an entity in a BoHV-1 infected MDBK cell (D: This figure has been reproduced with permission of P. Wild et al., Micron 33, 2002, Elsevier). Bars 200 nm.
Capsids are postulated to be enveloped at the trans Golgi network (Mettenleiter et al., 2006) by a process designated wrapping. However, capsids can bud at any location of the Golgi complex (Figure 9B) and vacuoles as have been shown for HSV-1 (Leuzinger et al., 2005; Stannard et al., 1996), BoHV-1 (Wild et al., 2002) and pseudorabies virus (Klupp et al., 2008), and even at microsomes (Albecka et al., 2016; Hollinshead et al., 2012). The result of wrapping is a small concentric vacuole (Figure 9D) containing a single virion as shown elsewhere in detail (Leuzinger et al., 2005; Wild et al., 2005). Golgi cisternae and vacuoles can contain one to numerous virions in a given section plane (Figure 9A). The cavities at the trans face of the Golgi complex in Figure 9A are more likely to represent cisternae rather than vacuoles because of their shape and location. The virions had gained access either by budding or by intraluminal transportation via ER-Golgi intermediates, as might be the case also in Figure 9C. Note that the viral envelope including spikes are covered by a dense layer in narrow Golgi cisternae (Figure 9C) whereas spikes are visible on virions in wide Golgi cisternae or large vacuoles (Figure 5 inset) and in the extracellular space (Figure 9E) as shown previously (Leuzinger et al., 2005; Wild et al., 2005). From the facts that virions are within ER and Golgi cisternae, and that the ONM continues via ER membranes into Golgi membranes forming an entity, we postulate that virions can be intraluminally transported from the PNS via ER into Golgi cisternae.
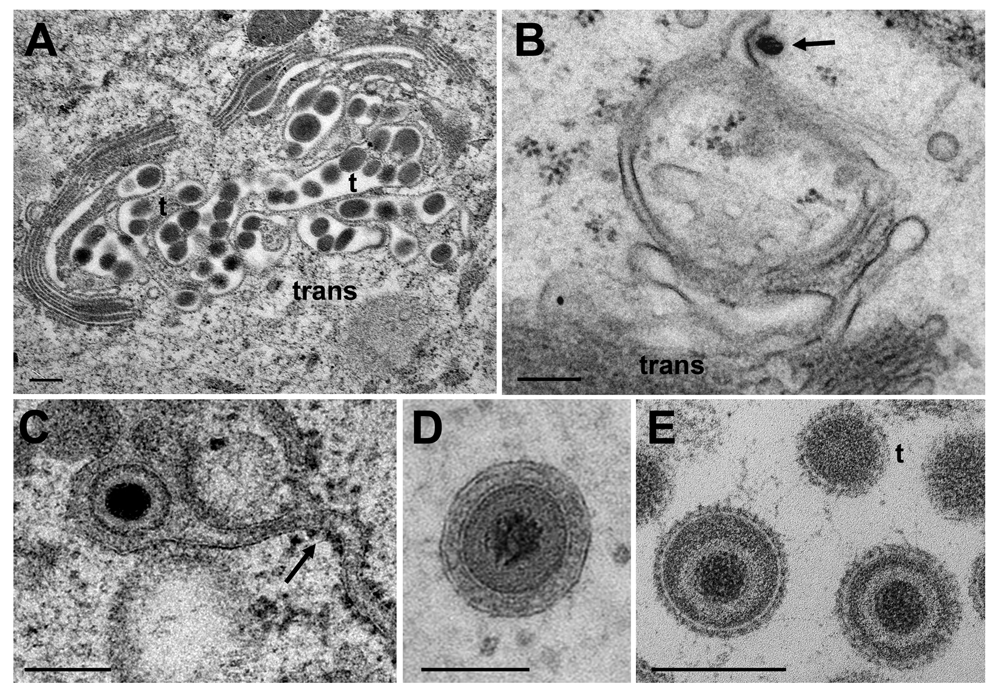
Many of them are tangentially (t) sectioned. (B) Budding BoHV-1 capsid at a Golgi membrane of the cis-face (arrow). (C) HSV-1 virion in a Golgi cisterna that connects to the ER (arrow). Note the dense content within the ER and Golgi cisterna indicating little loss of material during processing. (D) Concentric vacuole derived by wrapping containing a single BoHV-1 virion. The space between viral envelope and vacuolar membrane is always filled in well preserved cells. (E) Virions in a large vacuole or cisterna exhibiting clearly spikes even in tangentially (t) sectioned virions. Bars: 200 nm.
ER-to-Golgi transitions are not only established in infected cells, but also in cultured epithelial cells (Figure 10A) or in cells in organs, e.g. parathyroid gland, which was prepared by perfusion fixation (Wild et al., 1985) according to conventional protocols (Figure 10B). These observations suggest that ER to Golgi transitions may also serve as a direct transportation route e.g. for proteins to be finally released by exocytosis.
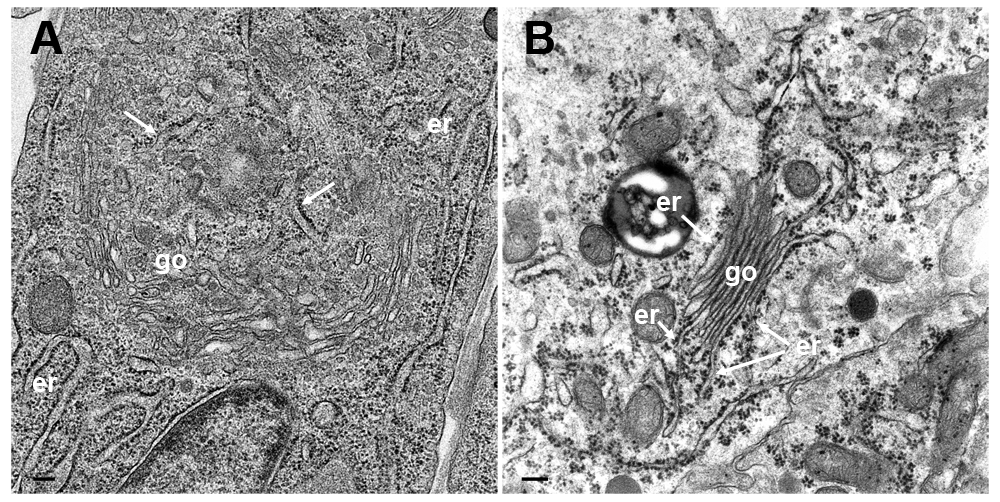
(A) TEM image of a cultured epithelial cell in which the Golgi complex is embedded in the ER. The ER membranes (arrows) run into the Golgi complex, close to a structure that probably represents a tangential section of the Golgi organizing center. (B) TEM image of a parathyroid cell prepared according to conventional protocols showing Golgi membranes (go) continuing into ER membranes (er). Bars: 200 nm.
The Golgi complex is among the first organelles that rapidly disintegrate during processing for electron microscopy after improper fixation and processing (Han et al., 2013; Wild et al., 2001). To minimize disintegration, we employed a technique that leads to improved retention of cellular material (Cope & Williams, 1969; Weibull et al., 1984), and to improved spatial and temporal resolution (Mueller, 1992). This is especially important for analytical studies of cells in which the Golgi complex is involved in highly dynamic processes such as packaging of proteins into granules (Ellinger et al., 2010; Orci et al., 1981; Wild et al., 1982) in the secretory pathway, or envelopment of capsids and vacuole formation (Figure 11) for delivery of hundreds of virions to the cell periphery (Leuzinger et al., 2005; Wild et al., 2015; Wild et al., 2002). Because of the difficulties in preservation of the Golgi ultrastructure (Ellinger et al., 2010; Han et al., 2013; Wild et al., 2001), its three-dimensional structure is poorly understood. Cryo-FESEM revealed the Golgi complex to be a complex tightly packed structure. The membrane of the outermost cisternae may completely cover the cis-face. TEM also revealed that the Golgi complex is embedded in the ER system, with multiple membrane connections forming a Golgi-ER entity.
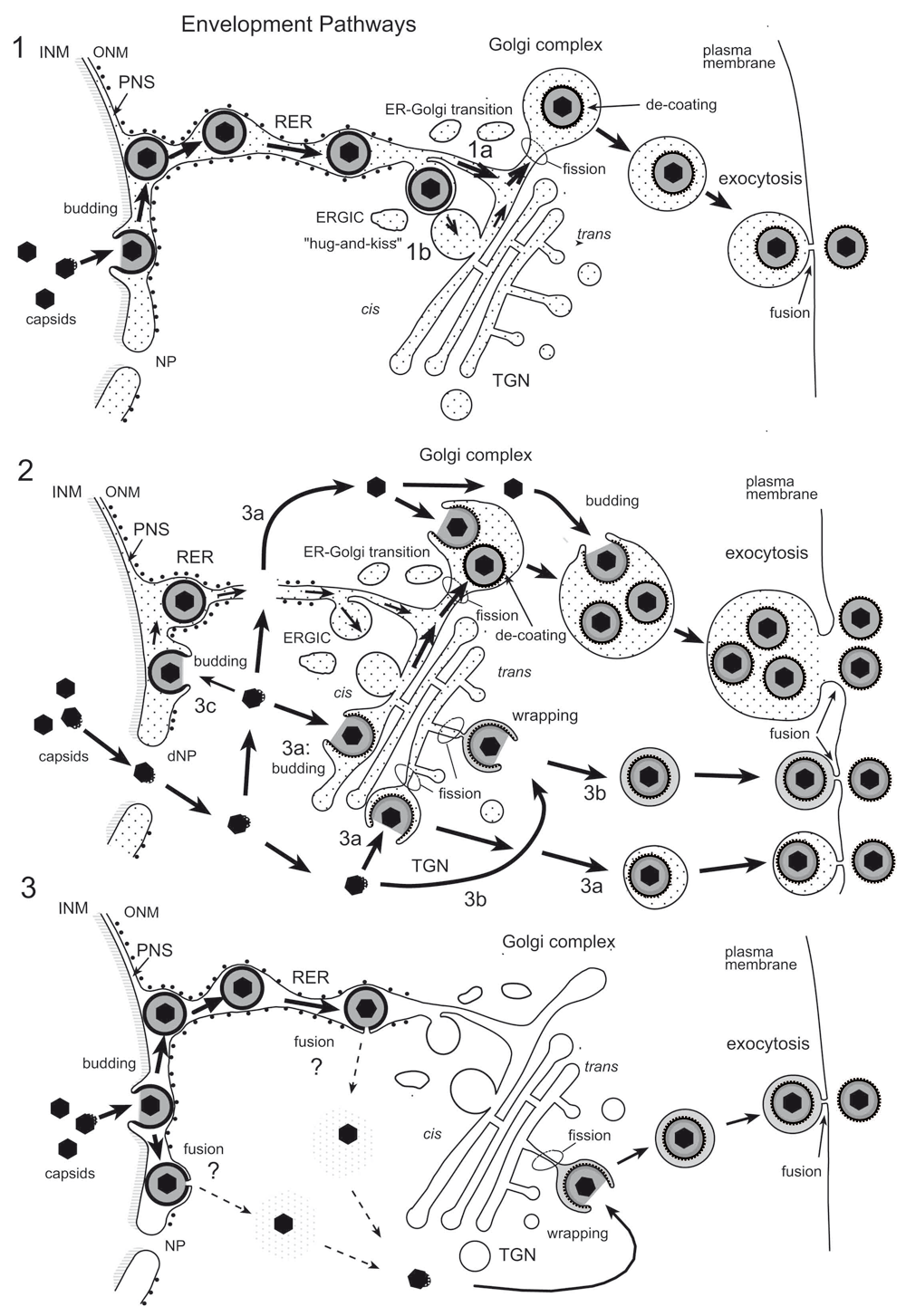
(1) Capsids bud at the INM into the PNS acquiring tegument and an envelope covered with a dense coat. These perinuclear virions are transported into the RER and further via Golgi transitions (1a) or the ERGIC (“hug-and-kiss”, 1b) into Golgi cisternae where they are packaged into transport vacuoles, which are detached from Golgi membranes by fission. The dense coat is shed off while vacuoles are transported to the cell periphery for exocytotic release of uncoated virions into the extracellular space. (2) Capsids gain direct access to the cytoplasmic matrix via dilated nuclear pores (dNP), and are transported to any site of the Golgi complex. They either bud into Golgi cisternae and vacuoles, respectively (2a) or are enveloped by a process designated wrapping (2b) that involves budding and concomitant formation of a small transport vacuole engulfing a single virion. Occasionally, capsids may bud at the OM or RER (2c), and the resulting virions are intraluminally transported as in pathway 1. (3) After budding at the INM, capsids and tegument of perinuclear virions are released into the cytoplasmic matrix via fusion of the viral envelopment (including dense coat) with the ONM (de-envelopment). Capsids are then re-enveloped at the trans Golgi network (TGN) by wrapping. Finally, vacuoles derived by fission from Golgi membranes transport virions to the cell periphery and release them into the extracellular space via exocytosis. The dense coat, which derived during the budding process at the INM and probably protects the viral envelope from fusion with membranes the virions are transported along, is shed of (de-coating) in transport vacuoles at latest when virions are released into the extracellular space. During budding at Golgi cisternae and vacuoles, a dense rim of tegument is closely attached to the inner layer of the viral envelope. No dense coat is formed so that spikes (glycoproteins) are readily seen in high resolution micrographs.
The Golgi complex fragments and disperses about 16 hpi with HSV-1 (Campadelli et al., 1993) adding additional difficulties for understanding Golgi function in herpes virus envelopment. To address the significance of the Golgi complex in virus envelopment and virus transportation, we thus investigated infected cells between 8 hpi (the approximate time of onset of envelopment) and 16 hpi. Our data clearly show that the Golgi complex is localized in a juxtanuclear position appearing as a compact entity by cryo-FESEM. Golgi membranes continue into ER membranes which in turn connect to the ONM forming a continuum between Golgi cisternae and PNS. Thus, the presence of virions within the PNS, ER cisternae and Golgi cisternae strongly suggests that the ER-to-Golgi transition is used as a direct intraluminal pathway to deliver virions from the PNS into Golgi cisternae (Figure 11, pathway 1). This idea is supported by the fact that about 80 HSV-1 virions per mean cell volume were within ER cisternae at 12 and 16 hpi but close to 300 by 24 hpi (Wild et al., 2015) suggesting that virus transportation out of the ER is inhibited after Golgi fragmentation (Campadelli et al., 1993). Virus transportation out of the ER is also drastically inhibited after BFA exposure. The ER delates and secretory protein transport from the ER to the Golgi complex is impeded after BFA treatment (Fujiwara et al., 1988; Misumi et al., 1986). Hence, the integrity of the Golgi complex is crucial for export of both secretory proteins and HSV-1 out of the ER suggesting that HSV-1 release from the ER to the Golgi complex follows a similar pathway as secretory proteins either by vesicle formation involving cop II (Klumperman, 2000) or equivalent, or via ER-Golgi transitions.
According to the currently stressed herpes virus egress theory (Figure 11, pathway 3), formation of infectious herpes viruses follows a complicated uneconomic pathway involving primary envelopment by budding of capsids at the INM, de-envelopment of capsids by fusion of the viral envelope with the ONM releasing capsids and tegument into the cytoplasmic matrix, and re-envelopment by wrapping at the trans Golgi network. The interaction of the viral envelope with the ONM was described the first time in 1968 (Darlington & Moss, 1968) and identified as budding of capsids from the cytoplasmic matrix into the PNS. About 30 years later, it was tried to prove that this process is fusion (Kopp et al., 2002; Naldinho-Souto et al., 2006; Skepper et al., 2001). Fact is that the phenotypes of the process taking place at the ONM are identical with those at the INM (Darlington & Moss, 1968; Leuzinger et al., 2005; Wild et al., 2005; Wild et al., 2012) and that they show all characteristics of budding. Budding requires proteins that are able to induce positive and negative curvatures. Budding at the INM is driven by UL31/UL34 (Bigalke & Heldwein, 2016; Bigalke & Heldwein, 2017; Bigalke et al., 2014; Hagen et al., 2015). UL31 and UL34 are also present at the ONM even in cells infected with Us3 deletion mutants (Reynolds et al., 2002) those envelopes are unable to fuse with the ONM (Reynolds et al., 2002; Wisner et al., 2009). Therefore, the presence of UL31/UL34 at the ONM cannot be the result of membrane transportation from the INM to the ONM via budding and subsequent fusion as often used as arguments for the presence of viral proteins at the ONM. Furthermore, UL34 was shown to localize at the ER (Yamauchi et al., 2001) and that UL31 is required for its dislocation to the INM. Recently, it was accidentally proved that the virus interaction at the ONM and ER membranes is budding rather than fusion. Glycoproteins B and H (gB/gH), members of the quartet responsible for cell entry (Turner et al., 1998), have been claimed to be responsible for fusion of the viral envelope with the ONM (Farnsworth et al., 2007). Electron microscopy strikingly revealed virus interactions with the ONM and ER membranes showing all characteristics of budding in cells infected with a gB/gH deletion mutant. Since the viral envelope cannot fuse in the absence of gB/gH the phenotypes shown in Figure 2 of this report (Farnsworth et al., 2007) represent undoubtedly various stages of capsids budding from the cytoplasmic matrix into the PNS and ER cisternae. Furthermore, in Vero cells infected with the gB/gH deletion mutant, three times more virions were found in the cytoplasm compared to wild type infected Vero cells, and about a third in the extracellular space. Similar relations though less pronounced were found in other cell types absolutely implying another transportation route than that of de-envelopment by fusion of the viral envelope with the ONM.
Wrapping demands an enormous amount of membranes that would need to be provided to the TGN, which, to our knowledge, is not clear yet. However, capsids can be wrapped at diverse sides of the Golgi complex or can bud into Golgi cisternae and/or vacuoles via another pathway than wrapping. There are numerous reports showing HSV-1 virions in vacuoles and/or Golgi cisternae (Homman-Loudiyi et al., 2003; Leuzinger et al., 2005; Stannard et al., 1996; Sutter et al., 2012; Wild et al., 2015; Wild et al., 2002). Budding of capsids at various sites of Golgi membranes as well as virions in Golgi cisternae and vacuoles were also shown in pseudorabies virus infected cells (Klupp et al., 2008). These virions in all these vacuoles and/or Golgi cisternae did not arise by wrapping because wrapping results in a single virion in a concentric vacuole. They may have entered Golgi cisternae by budding into them or by intraluminal transportation via ER-to-Golgi transitions. Transient elements from cis and trans Golgi sides have been shown in various cells (Pavelka & Roth, 2015). It was also suggested that the cis-Golgi approaches the ER and contacts the ER exit sites in the yeast Saccharomyces cerevisiae to capture cargo for transportation to the Golgi complex (Kurokawa et al., 2014). This ‘hug-and-kiss’ behavior could be another route to transfer virions, which are intraluminally transported to ER exit sites, into the Golgi cisternae.
In cells prepared for improved resolution, we found no valid arguments for de-envelopment by fusion of the viral envelope with the ONM. There are a number of facts that argue clearly against the de-envelopment theory. First, the morphology of the process taking place at the INM and ONM are identical showing all characteristics for budding (Darlington & Moss, 1968; Leuzinger et al., 2005; Wild et al., 2005; Wild et al., 2012) but none for fusion (Haluska et al., 2006; Kanaseki et al., 1997) considering fundamentals of membrane bound transportation (Leabu, 2006; Peters et al., 2004; White, 1992). Second, virions have been repeatedly shown within ER cisternae (Gilbert et al., 1994; Granzow et al., 1997; Leuzinger et al., 2005; Radsak et al., 1996; Schwartz & Roizman, 1969; Stannard et al., 1996; Sutter et al., 2012; Wild et al., 2002). To reach this location, either virions need to be transported from the PNS into the ER, or capsids have to bud from the cytoplasmic matrix into the ER. If virions can be intraluminally transported out of the PNS the viral envelope must be protected from fusion with membranes the virions are transported along. If capsids have the ability to bud at ER membranes capsids are very likely to be able to bud at the ONM since the ONM is part of the ER. Third, virions can accumulate to large numbers in the PNS, e.g. in the absence of the protein kinase Us3 (Poon et al., 2006; Reynolds et al., 2002; Wild et al., 2015). Strangely enough, these virions are infective (Reynolds et al., 2002; Ryckman & Roller, 2004; Wild et al., 2015) despite the inability of the envelope to fuse with the ONM (Wisner et al., 2009) clearly contradicting the theory that de- and re-envelopment is essential to become infective (Klupp et al., 2011). Fourth, the equivalent to budding is the formation of coated pits resulting in coated vesicles (Owen & Luzio, 2000; Pearse et al., 2000). Coated vesicles derive e.g. from the plasma membrane and are transported towards the Golgi complex where they fuse with Golgi membranes (Orci et al., 1981). However, coated vesicles must be uncoated to gain the ability for fusion. The coat consists of clathrin that drives formation of coated pits and finally coated vesicles and protects them from fusion. Clathrin was claimed to be involved in envelopment of HSV-6 capsids at Golgi membranes together with viral proteins (Mori et al., 2008). Budding of HSV-1 capsids at the INM is driven by the nuclear envelopment complex, UL31/UL34, (Bigalke & Heldwein, 2015; Bigalke et al., 2014; Hagen et al., 2015) located at the nuclear rim (Mou et al., 2009).
Budding at the INM, ONM and ER membranes starts with deposition of dense substances that finally result in a dense coat at the viral envelop. The dense coat is readily seen on virions in the PNS, ER and, inconsistently, in Golgi cisternae and large vacuoles containing many virions. The dense coat suggests that it protects virions from fusion with membranes the virions is transported along but allows virus transportation from the PNS into ER and Golgi cisternae. However, in large Golgi cisternae and/or vacuoles, many virions are without dense coat. Instead, spikes are visible like at virions in the extracellular space. Budding at Golgi membranes takes place without dense coat formation.
Golgi membranes interconnect with ER membranes in cells infected with HSV-1, a Us3 deletion mutant thereof or with BoHV-1 as well as in uninfected cells. The ER continues into the ONM, that turns into the INM at sites of nuclear pores. Consequently, the PNS, ER and Golgi complex forms an entity that can be only visualized by TEM, either when the membranes of all three compartments are luckily hit in the same plane of a given section, or by 3D-reconstruction after imaging of serial sections, focused ion beam (FIB) microscopy or electron tomography. Fact is that virions are intraluminally transported out of the PNS into the ER. This implies that the viral envelope needs to be protected from fusion with the membranes the virions are transported along. Thus, the significance of the dense coat, which derives during budding of capsids at nuclear membranes, is protecting the viral envelope from fusion in a similar manner as clathrin protects coated vesicles from fusion. The ER-to-Golgi transitions, virions in Golgi cisternae with a similar dense coat as virions in the PNS and ER, and inhibition of virion transportation out of the PNS and ER after disruption of the Golgi complex by BFA, strongly suggest that virions are intraluminally transported from the PNS through the ER into Golgi cisternae. We propose that this is the only pathway for virions out of the PNS, and that capsids gain direct access to the cytoplasm from the nucleus via dilated nuclear pores and impaired nuclear envelope.
Dataset 1: Raw images for Figure 1–Figure 5, Figure 8–Figure 10. DOI, 10.5256/f1000research.12252.d179644 (Wild et al., 2017a)
Dataset 2: Raw values for Figure 6 and Figure 7. DOI, 10.5256/f1000research.12252.d179645 (Wild et al., 2017b)
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)