Keywords
Protein evolution, protein structure, evolutionary rate, relative solvent accessibility, weighted contact number, multiple sequence alignment
Protein evolution, protein structure, evolutionary rate, relative solvent accessibility, weighted contact number, multiple sequence alignment
Different sites within a protein-coding gene evolve at different rates1,2. This evolutionary rate heterogeneity across protein sites results from a complex interplay of both functional and structural constraints3. For example, residues that are critical to a given protein’s function, such as residues involved in enzymatic activity, in protein–protein interactions, or in protein–ligand interactions, tend to evolve more slowly than other residues in the protein4–10. In addition, protein structure has been found to play a major role in shaping protein evolutionary rates across the entire protein sequence, because the imperative for a protein to stably fold produces an overarching evolutionary constraint. Structurally-important protein residues (namely residues in the protein core) tend to be highly conserved and evolve very slowly, but residues with a minor influence on structure (namely surface residues) evolve more rapidly4,9,11–19.
To study evolutionary conservation in a structural context, we need methods to (i) measure evolutionary rates at individual sites in a protein alignment, (ii) map those rates onto the protein structure, and (iii) quantify site-level structural properties. Here, we describe in detail how to perform these three steps, considering a few commonly used alternatives at both steps (i) and (iii). In addition, we provide extensive notes highlighting specific technical issues and/or describing alternative analysis approaches. At step (i), we demonstrate how evolutionary rates can be measured using either amino-acid or codon data. For amino-acid data, we consider relative amino-acid rates, i.e., rates of evolutionary variation normalized by the mean of the rate in the entire protein10,20. For codon data, we consider site-specific dN/dS values. These are site-specific rates of nonsynonymous variation normalized by (whole-gene) rates of synonymous variation21,22. At step (iii), we discuss two related but somewhat distinct structural measures. First, we consider the solvent accessibility, which measures the extend to which a site is exposed to the solvent environment. Specifically, we consider the relative solvent accessibility (RSA)23, which is the solvent accessibility of a residue in a structure normalized by the maximally possible solvent accessibility of that residue in a Gly-X-Gly tripeptide. Second, we consider the packing density, which measures the proximity and number of neighboring residues. Specifically, we consider the side-chain weighted contact number (WCN)19, which is calculated relative to the geometric center of the residue side-chain atoms and employs an inverse-square weighting term.
Below we list the software packages needed to perform the analysis. Please download the most recent version of each software, unless a specific version is specified in the text. The links provided contain instructions for installing and testing the software.
1. HyPhy (see Note 1)
HyPhy is a general-purpose software platform for inference in a phylogenetic framework24. To install, clone the HyPhy git repository to your desired directory. The HyPhy repository can be found at https://github.com/veg/hyphy.git. Instructions for installation are available from http://hyphy.org/installation.
2. MAFFT
MAFFT is a program for generating multiple sequence alignments25. Download MAFFT from http://mafft.cbrc.jp/alignment/software/.
3. RAxML
RAxML is a tool for phylogenetic inference using maximum likelihood26. Clone the RAxML repository to a local directory. The RAxML git repository can be found at https://github.com/stamatak/standard-RAxML. Analyses presented here utilize the raxmlHPC-SSE3 executable, which can be compiled with Makefile.SSE3.gcc or Makefile.SSE3.mac. Note that this executable does not allow threading. (See Note 2 for information on how to enable threading.)
4. mkDSSP
mkDSSP is a tool that calculates solvent accessibilities and parses secondary structure assignments from a PDB input file into a standardized format27. This format follows that of the entries in the DSSP database28. Download the mkDSSP software from https://slackbuilds.org/repository/14.2/academic/mkDSSP/.
5. Python
Download Python from https://www.python.org/downloads/.
6. Biopython
Biopython is a python library for computational molecular biology29. Download Biopython from http://biopython.org/wiki/Download.
Biopython has several dependencies that also need to be installed. You can find the information about installing the dependencies in the link provided.
7. argparse
argparse is a python module providing user-friendly command-line interfaces. We use argparse in most of our custom python scripts. Install argparse using the link https://pypi.python.org/pypi/argparse.
8. pandas
pandas is a python module for data manipulation and analysis. You can download pandas from https://pandas.pydata.org/getpandas.html.
9. R
Download R from https://www.r-project.org/. We recommend to use RStudio to execute and edit R scripts. RStudio can be installed from https://www.rstudio.com/. We will use R for data visualization. Our scripts require the packages dplyr, readr, cowplot, and their dependencies. You can install an R package by typing the command install.packages("dplyr") (for installing dplyr) in the R shell. By default, this command will also install any dependencies needed for the package to work.
10. Custom scripts (see Note 3)
All our custom python, R, and HyPhy scripts can be found at: https://github.com/clauswilke/proteinER/tree/master/src.
In the following, we provide four separate protocols to (i) measure relative amino acid rates, (ii) measure site-specific codon evolutionary rates (expressed via the metric dN/dS), (iii) measure structural quantities such as RSA and WCN, and (iv) combine the measured quantities into a combined analysis. To provide an example, we demonstrate all four protocols on an empirical dataset consisting of mammalian orthologs of histamine receptor 1 (ENST00000438284) and an accompanying PDB structure. This dataset was originally analyzed by Spielman and Wilke30. Throughout, we assume that we are working on a UNIX-like command line interface. For your convenience, we have provided a git repository at https://github.com/clauswilke/proteinER/ that contains the input and output files used in each step. Our overarching strategy throughout this work is to first infer a given measurement (e.g., dN/dS or RSA) for each site in the multiple sequence alignment or protein structure. To compare the different measurements, we then map them all to columns in the multiple sequence alignment.
The input and output files used in this section can be found at: https://github.com/clauswilke/proteinER/tree/master/measuring_aa_rates.
1. Align sequences with MAFFT
Store all of the sequences you wish to align into one file. The file must be in the FASTA format. The FASTA format contains two pieces of information for each sequence: the sequence ID preceded by a ">" sign and followed by a new line, and then the sequence itself. We will use the FASTA file HRH1_unaligned.fasta that contains homologous sequences that are not aligned. We align them with the command:
mafft --auto --inputorder \ HRH1_unaligned.fasta > \ HRH1_aligned.fasta
Arguments above correspond to the following:
• --auto, Select the optimal alignment algorithm for the given data.
• --inputorder, Output sequences in the same order in which they were provided. Without this option, the order of the sequences in the alignment is arbitrary.
The output file HRH1_aligned.fasta will contain the aligned sequences.
2. Infer tree with RAxML (see Notes 2, 4)
Using the file with the alignment HRH1_aligned.fasta, run RAxML with the following command:
raxmlHPC-SSE3 -s HRH1_aligned.fasta \ -n HRH1_tree \ -m PROTCATLG \ -p 12345
Arguments above correspond to the following:
• -s, The multiple sequence alignment file.
• -n, The extension for the outputted tree files. Here, the outputted files will contain HRH1_tree in their names.
• -m, The model of sequence evolution, in this case the LG amino-acid model31 with RAxML’s “CAT" model32 of sequence heterogeneity.
• -p, The random number seed initializing this phylogenetic inference. To reproduce the exact phylogeny we have, specify this random seed.
The desired tree file is RAxML_bestTree.HRH1_tree.
3. Infer site-wise rates with HyPhy (see Note 5)
To run HyPhy, the file runRelativeProtRates.bf must be edited to specify the directories and file names that will be used in the analysis. Edit these two lines of runRelativeProtRates.bf
"0": "/path/to/HRH1_aligned.fasta", "1": "/path/to/RAxML_bestTree.HRH1_tree",
Here, "0" specifies the full path to the alignment file HRH1_aligned.fasta, and "1" specifies the full path to the tree file RAxML_bestTree.HRH1_tree.
Run HyPhy with the command
HYPHYMP runRelativeProtRates.bf
An output file HRH1_aligned.fasta.site-rates.json is written to the folder that contains the alignment.
4. Parse HyPhy output (see Note 3)
For further downstream processing, the HyPhy output file in JSON format needs to be converted to CSV format. The custom python script parse_prot_rates.py will extract the site’s position, rate, and other site information outputted by HyPhy. Parse the JSON file with the command
python parse_prot_rates.py \ -j HRH1_aligned.fasta.site-rates.json \ -r HRH1_rates.csv
Arguments above correspond to the following:
5. Calculate relative site-wise rates
As discussed by Jack et al.10, we recommend calculating relative evolutionary rates by normalizing inferred site-specific rates by their average. In other words, to compute the relative amino-acid rates, calculate the mean rate of the entire sequence and divide each site’s rate by this mean rate. Once normalized, a rate below 1 will indicate a site that evolves more slowly than average. For example, a rate of 0.5 implies that the corresponding site evolves half as fast as the average. Similarly, a rate above 1 will indicate a site that evolve more quickly than average. For example, a rate of 2 implies that the corresponding site evolves twice as fast as the average.
The input and output files used in this section can be found at: https://github.com/clauswilke/proteinER/tree/master/measuring_dNdS.
1. Translate codon sequences (see Note 3)
In this section, both codon and amino-acid sequences are required to perform site-wise rate calculations. Store all of the desired nucleotide sequences into one FASTA file. Use our custom script to convert a codon FASTA file to an amino acid FASTA files. We use the FASTA file HRH1_unaligned_codon.fasta that contains homologous nucleotide sequences we wish to translate. Translate with the command:
python translate_aln_codon_to_aa.py \ -n HRH1_unaligned_codon.fasta \ -o HRH1_unaligned_aa.fasta
Arguments above correspond to the following:
2. Align amino acid sequences with MAFFT
Align amino acid sequences using step 1 in Protocol 1.
3. Back-translate the amino acid alignment into a codon alignment (see Note 3)
This step requires the original codon sequences and the amino acid alignment. Note that the amino acid alignment is retained, and the script simply inserts corresponding codons in place of amino acids at each column of the alignment. The command to back-translate the sequences is:
python translate_aln_aa_to_codon.py \ -a HRH1_aligned_aa.fasta \ -n HRH1_unaligned_codon.fasta \ -o HRH1_aligned_codon.fasta
Arguments above correspond to the following:
4. Infer tree with RAxML
The following step is the same as step 2 in Protocol 1. Use the amino-acid alignment file HRH1_aligned_aa.fasta to infer the tree.
5. Infer site-wise rates with HyPhy (see Note 6)
To run HyPhy, the file runFEL.bf must be edited to specify the directories and file names that will be used in the analysis. Edit the following two lines of the runFEL.bf script:
"1": "/path/to/HRH1_aligned_codon.fasta", "2": "/path/to/RAxML_bestTree.HRH1_tree",
Here, "1" specifies the full path to the alignment file HRH1_aligned_codon.fasta, and "2" specifies the full path to the tree file RAxML_bestTree.HRH1_tree.
Run HyPhy with the following command:
HYPHYMP runFEL.bf
An output file
HRH1_aligned_codon.fasta.FEL.json is written to the folder that contains the alignment file.
6. Parse HyPhy output (see Note 3)
For further downstream processing, the HyPhy output file in JSON format needs to be converted to CSV format. The custom python script parse_FEL.py will extract the site’s position, dN/dS, and other site information outputted by HyPhy:
python parse_FEL.py \ -j HRH1_aligned_codon.fasta.FEL.json \ -r extracted_HRH1_dNdS.csv
Arguments above correspond to the following:
7. Change dN/dS values for conserved sites (see Note 3)
The FEL method as implemented in HyPhy assigns dN/dS = 1 to sites without any synonymous and any non-synonymous substitutions. We recommend to express rate at entirely conserved sites with dN/dS = 0. We provide a custom script that will assign dN/dS = 0 to completely conserved sites. This script will not change extracted_HRH1_dNdS.csv’s original format.
python fix_dNdS_conserved_sites.py \ -a HRH1_aligned_aa.fasta \ -r extracted_HRH1_dNdS.csv \ -o processed_HRH1_dNdS.csv
Arguments above correspond to the following:
All structural features in this section are calculated from an example PDB file, 3rze.pdb. This PDB file defines the crystal structure of a transmembrane protein fused to an unrelated lysozyme protein. The lysozyme is required for crystallization, but is not biologically relevant. We have pre-processed the PDB file to exclude residues from the lysozyme protein (residue numbers 1000 and above). The input and output files used in this section can be found at: https://github.com/clauswilke/proteinER/tree/master/measuring_structural_features.
1. Calculate relative solvent accessibility (RSA) from the PDB file (see Note 3)
We provide a custom script calc_rsa.py that will run mkdssp27,28, extract absolute solvent accessibilities, and calculate relative solvent accessibilities23. The first argument is the PDB file, and the second optional argument (-o 3rze) is the prefix used for the output files.
python calc_rsa.py 3rze.pdb -o 3rze
This command will generate two output files: 3rze.asa.txt containing the raw mkdssp output, and 3rze.rsa.csv containing RSA values and secondary structure classifications.
2. Calculate weighted contact numbers (WCN) from the PDB file (see Note 3)
WCN measures amino acid packing density, and may be calculated with respect to either the α-carbon or the geometric center of the side-chain33,19. We provide a custom script that will calculate both types of WCN values. The command line arguments follow the same format as the calc_rsa.py script.
python calc_wcn.py 3rze.pdb -o 3rze
The above command will produce an output file 3rze.wcn.csv that contains both side-chain WCN and α-carbon WCN values for each position in the input PDB file.
The input and output files used in this section can be found at: https://github.com/clauswilke/proteinER/tree/master/map_structural_features.
1. Generate sequence alignment map (see Note 3)
To map site specific evolutionary rates to residues in a PDB structure, we first align the sequence of amino acids extracted from the PDB structure to the multiple sequence alignment used for rate inference. We provide a script that calls mafft to align a PDB sequence to a multiple sequence alignment and reformat the output.
python make_map.py \ HRH1_aligned.fasta 3rze.pdb
Running the above command produces a CSV file with four columns. The first column contains, for each residue, the numbered position of that residue in the alignment used for rate inference. The second column similarly contains the numbered position of each residue in the PDB structure. Numbered positions are extracted directly from the PDB input file and may include PDB insertion codes (see Note 8). The third and fourth columns contain the single-letter amino acid present in the PDB structure and the PDB chain, respectively. If an amino acid is in the alignment but not in the PDB structure, the PDB position is assigned a value of NA. Likewise, if an amino acid is in the PDB structure but not the alignment, the alignment position is assigned NA.
2. Map rates to structural features (see Note 3)
After mapping the alignment used for rate inference to the sequence of the PDB structure, we align rates with structural features. We provide a script that uses the map generated above to combine rates and structural features into a single CSV.
python map_features.py 3rze.map.csv \ -r processed_HRH1_dNdS.csv \ HRH1_rates.csv \ -f 3rze.rsa.csv 3rze.wcn.csv
Arguments above correspond to the following:
• The input file containing a map between the alignment residue positions and the structure residue positions.
• -r, The rates files.
• -f, The structural feature files.
• -o, The CSV output file. If not specified, the script outputs a file <pdb_id>.rates_features.csv. Here, <pdb_id> is the name of the PDB ID used to make the map file.
The output from this command provides all the data needed to compute correlations between rates and structural features and corresponding visualizations. See Figure 1 as an example.
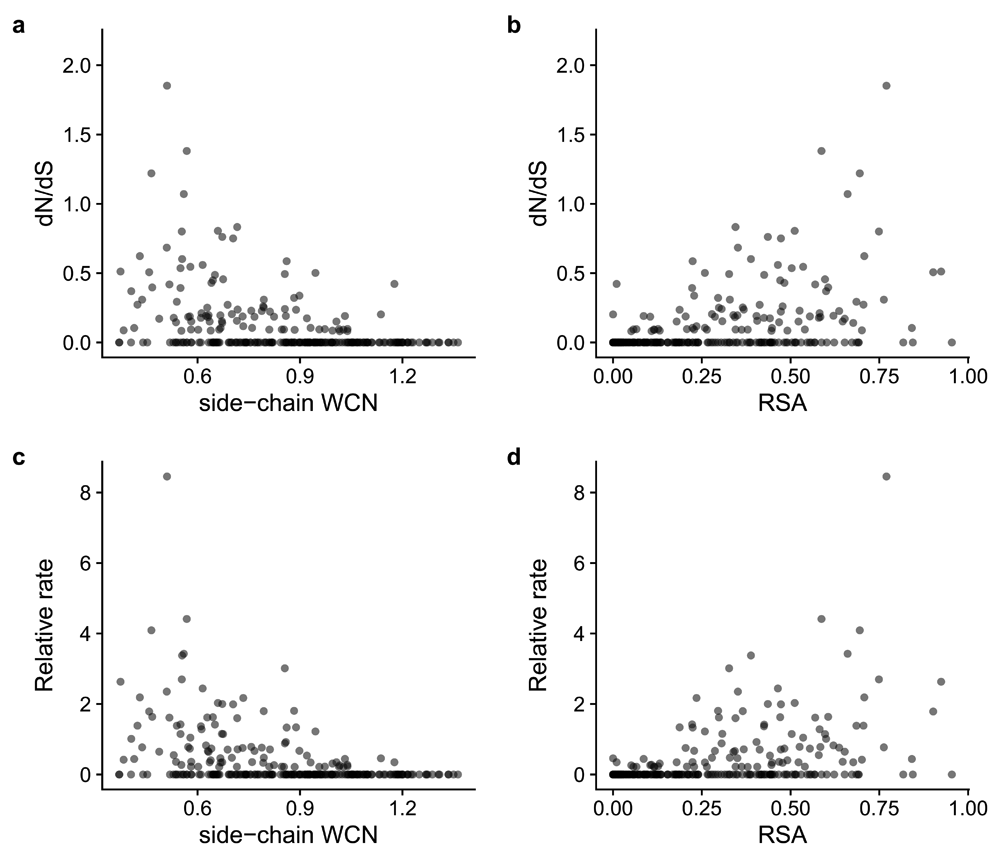
(a–d) Each point represents a residue in the structure of the HRH1 protein (PDB: 3rze). The Pearson correlation coefficients r between structural features (RSA or WCN) and rates (dN/dS or amino acid) are as follows for each panel: (a) –0.39, (b) 0.43, (c) –0.39, (d) 0.42.
We have provided four separate protocols that jointly enable the analysis of protein evolutionary rates in a structural context. The first two protocols are used to measure site-specific evolutionary rates from multiple-sequence alignments, either at the amino-acid or the codon level. Any actual study will generally employ only one of these protocols. The third protocol is used to quantify local characteristic of a protein structure, such as relative solvent accessibility or weighted contact number, and the fourth protocol maps the structural quantities to the evolutionary rates and vice versa. We hope that these protocols will be useful for further research into disentangling structural and functional constraints on protein evolution.
1. The minimum required HyPhy version for FEL dN/dS inference is 2.3.3. The minimum required version for relative amino-acid rate inference is 2.3.4.
2. To thread RAxML, compile the raxmlHPC-PTHREADS-SSE3 executable with Makefile.SSE3.PTHREADS.gcc or Makefile.SSE3.PTHREADS.mac. The options to call RAxML stay the same. Add the option -T to thread, and run RAxML with
raxmlHPC-PTHREADS-SSE3 -T 48 \ -s HRH1_aligned.fasta \ -n HRH1_tree \ -m PROTCATLG \ -p 12345
3. All of our custom python scripts provide documentation when called with the options -h or --help. For example, to view the documentation for the script calc_rsa.py, run the command
python calc_rsa.py -h
The script’s use and required input files will be described in the documentation. Additionally, where applicable, the documentation also provides a description of the information stored in the output files.
4. RAxML can also infer trees from nucleotide sequence data in addition to amino-acid data. Importantly, if the analyzed sequences are highly diverged, trees inferred from nucleotide sequences may yield better rate predictions. To infer a tree from nucleotide data with RAxML, issue the following command (specifically, -m PROTCATLG has been changed to a GTR nucleotide model with CAT heterogeneity, -m GTRCAT):
raxmlHPC-SSE3 -s HRH1_aligned.fasta \ -n HRH1_tree -m GTRCAT \ -p 12345
Furthermore, if the dataset of interest contains fewer than 50 taxa, it is not recommended to adopt the CAT model of heterogeneity32. Instead, a discrete Gamma distribution should be used. To specify this model, simply replace the phrase CAT with GAMMA: For amino-acids, use the model specification -m PROTGAMMALG, and for nucleotides use the model specification -m GTRGAMMA.
5. As an alternative method to infer site-wise amino acid rates one can use Rate4Site. Rate4Site is a tool for inferring site-wise evolutionary rates in amino acid sequences20. Download Rate4Site from https://www.tau.ac.il/~itaymay/cp/rate4site.html. Analyses presented here use Rate4Site downloaded as rate4site.3.2.source.zip and compiled with the Makefile_slow file.
The options to run Rate4Site may be different for different Rate4Site installation files. We recommend using the rate4site -h command to find the proper options for your version, as opposed to using the software’s website.
Run the following command to infer site-wise rates:
rate4site -Mw -s HRH1_aligned.fasta \ -t RAxML_bestTree.HRH1_tree \ -o HRH1_norm_rates.txt \ -y HRH1_orig_rates.txt
Arguments above correspond to the following:
• -Mw, Specify the WAG model of amino-acid evolution (see Note 7).
• -s, The multiple sequence alignment file.
• -t, The input phylogeny.
• -o, The output file of normalized amino-acid rates.
• -y, The output file of raw amino-acid rates.
Rate4Site normalizes rates by converting them into z-scores. The z-scores are written to HRH1_norm_rates.txt. Rate4site also outputs the raw (unnormalized) scores in HRH1_orig_rates.txt. We advise you to use raw scores and to normalize them by the average score in the sequence, as discussed in protocol 1 step 5. Note that Rate4Site also outputs a new tree file TheTree.txt and an empty rates file r4s.res. These files are not needed for further analysis.
For further downstream processing, the Rate4Site output file needs to be converted to a CSV file. The following command will extract the site’s position, amino acid, and Rate4Site score (see Note 3).
python parse_r4s.py \ HRH1_orig_rates.txt \ -o extracted_HRH1_orig_rates.csv
Arguments above correspond to the following:
By default, the Rate4Site software will output rates for the sites in the first sequence of the alignment file. That is, a gap in the first sequence will not be assigned a rate, even though a rate is inferred for every site in the alignment. To circumvent losing information outputted from Rate4Site, we suggest finding the sequence in the alignment with least amount of gaps and using it as the reference sequence for the output. The reference sequence for Rate4Site can be specified with the option -a sequence_ID, where sequence_ID is the name of the sequence in a FASTA file provided for rate inference.
6. The file runFEL.bf implements fixed-effect likelihood (FEL) inference without synonymous rate variation, which is sometimes referred to as a one-rate FEL model. The model infers one dN value per site and one dS value per the entire sequence21. The one-rate FEL model has been found to infer more accurate dN/dS values than other HyPhy methods22.
7. For reasons that are beyond the scope of this paper, it turns out that the specific matrix choice has little effect on the final rates, as long as rates are normalized relative to their means as we do here. The underlying reason for this insensitivity to matrix choice is that the available matrices were all derived by pooling data from many sites in many proteins (see e.g.31), and this pooling yields matrices that are close to uninformative34,35.
8. The residue numbers in PDB files are not strictly sequential or numeric. If multiple residues share the same numeric value, they will be distinguished by a single letter insertion code (e.g. 53A or 53B)36. These insertion codes appear when there are several homologous proteins with crystal structures. Generally, each new structure retains the numbering of the earliest crystalized structure to preserve the alignment among structures of homologous proteins. If the new structure contains deletions relative to the original structure, the PDB file will skip residue numbers. If the new structure contains insertions, the PDB file will have residue numbers with insertion codes.
All information required to reproduce the analysis is provided at https://github.com/clauswilke/proteinER. Version 1.0 of this code is archived at https://doi.org/10.5281/zenodo.100594237.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)