Keywords
Dyslipidemia, Polymorphisms, HindIII, Lipoprotein Lipase, coronary artery disease
Dyslipidemia, Polymorphisms, HindIII, Lipoprotein Lipase, coronary artery disease
The relationship between dyslipidemia and atherosclerosis continues to be an area of active research, since the prevalence of atherosclerosis and associated cardiovascular complications continue to increase in the industrialized world1. Cardiovascular disease (CVD) constitutes the greatest cause of morbidity and mortality globally with a high incidence in countries of all economic categories2. Evidence supporting a causal relationship between lipid profile abnormalities and the risk of coronary artery disease (CAD) is overwhelming, confirming that hypercholesterolemia is an independent risk factor for CVD3–5. In addition, hypertriglyceridemia and mixed dyslipidemias have been associated with the aggregation of metabolic risk factors, like hypertension (HTN)6 and obesity7.
Dyslipidemias are a group of metabolic derangements characterized by any or a combination of the following: elevated low density lipoprotein (LDL-c) (>130md/dL), elevated total cholesterol (>200 mg/dL), elevated TG (>150mg/dL), or low high density lipoprotein (HDL-c) (<40mg/dL in men and <50mg/dL in women)8.
The worldwide prevalence of dyslipidemia varies between different individuals, depending on race, age, socio-economic and cultural factors, lifestyle and genetics. This prevalence has increased significantly in growing cities with economic growth9. According to the National Health and Nutrition Examination Survey (NHANES) 2003–2006, 53.0% of the adult population in United States has some form dyslipidemia10; however, a lower prevalence have been reported for other countries, for example Canada and South Korea, with 45% and 44.1%, respectively8,11. De Souza et al. studied a sample of 1,039 patients and reported that the most common dyslipidemias in Brazil were isolated low HDL-c (18.3%), hypertriglyceridemia (17.1%), and isolated hypercholesterolemia (4.2%)12. These results are similar to those reported by Aguilar-Salinas et al., in which the incidence of dyslipidemia in a group of 4,040 Mexican patients was 60.3%, from which low HDL-c represented 60.3%, hypercholesterolemia 43.6% and hypertriglyceridemia 31.5%13. Another cross-sectional and descriptive study carried out in 318 patients from Cuenca, Ecuador, found that 82.4% of individuals (86.8% in females and 76.5% in males) had some type of dyslipidemia. Isolated low HDL-c was the most prevalent abnormality and it was significantly associated with obesity (OR: 3.99. CI: 95%. 1.65-9.36; p<0.01)14.
In Venezuela, the CARMELA study evaluated the prevalence of lipid metabolism disorders in the city of Barquisimeto of Lara state, reporting one of the highest percentages in the country, with a 50.4% prevalence of dyslipidemia in this population15. Nevertheless, a study by Linares et al.16 with a sample of 2,230 individuals from Maracaibo City, Venezuela, identified the overall dyslipidemia prevalence was even higher at 84.8% (n=1892), where 88% of females and 81.4% of males were found to have dyslipidemia. High LDL-c was the most frequent abnormality found in this population (20%), followed by the combination of low HDL-c with high LDL-c (19%) and hypertriglyceridemia with high LDL-c and low HDL (16.2%). Bermúdez et al.17 found that low HDL-c was statistically associated with obesity, ethnic group, alcohol consumption, and elevated TG.
The association between family history of dyslipidemia and the risk of CVD is supported by a large body of evidence18–22. Additionally, the great advancement in DNA analysis techniques has aided research surrounding CVD and related genetics and epigenetics. Understanding gene mutations or polymorphisms involved in the synthesis, transport, and metabolism of lipoproteins allows recognition of potential therapeutic targets and alternative treatments through identification of new molecules1,3,20.
Dyslipidemia is one of the most well characterized cardiovascular risk factors19,20. This not only depends on diet, but also on the synthesis and metabolism of lipoproteins conditioned by gene expression. Given the importance and the great diversity of proteins that participate in lipid metabolism, one might expect that a single defect in any step of gene expression would affect the quantity or quality of the product and potentially predispose to dyslipidemias and CVD19.
One genetic abnormalities associated with low HDL-c and increased CVD risk is the Taq IB polymorphism located in chromosome 16q21. This gene alters cholesteryl ester protein transferase (CEPT), which decreases HDL-c concentration23. Some deletions, inversions, and substitutions of the APO AI-IV, CII, and CIII genes are also associated with both premature CVD and low HDL-c24,25. Total deficiency of lecithin cholesterol acyl transferase (LCAT) can be seen after transition of C→T in codon 147 of exon 4 (W147R), G→A in codon 293 of exon 6 (M293I), as well as partial deficiencies of LCAT due to transition of C→T. Additionally, the substitution of threonine for isoleucine in codon 123 (T123I) causes decreased HDL-c and higher cholesterol in the intima of arterial vessels26,27.
Below, some of the genetic alterations associated with low levels of HDL-c and a higher risk of CVD are highlighted:
1. CETP. The transcript of this gene mediates the exchange of lipids between lipoproteins, resulting in a net transference of cholesteryl esters from HDL to other lipoproteins and the capture of cholesterol in the liver. High levels of CETP lead to HDLs rich in TG, making a substrate for hepatic lipases, so that TG are hydrolyzed and ApoA-I is degraded in renal tubule cells. The subsequent decrease in HDL-c concentration creates a pro-atherogenic environment28. This occurs when CETP reaches a high level of expression in some individuals with polymorphisms in the codifying CETP gene (16q21). Being the most frequently occurring and best characterized polymorphism on intron 1, TaqIB is associated with the development of early atherosclerosis23.
2. Familial hypoalphalipoproteinemia and HDL-C deficiency. Approximately 50% of HDL-c alterations are explained by polygenic defects in various chromosomal loci that control apolipoprotein expression (A-I, A-II, C-II, C-III and Apo A-IV) and LCAT. Multiple genetic defects have been reported, such as deletions, inversions and substitutions on codifying genes of apolipoproteins, which are all associated with premature arterial disease24,25.
3. LCAT. This liver-synthesized enzyme circulates in plasma forming complexes with HDL and participating in the inverse transport of cholesterol. An LCAT deficiency results in the accumulation of free cholesterol on tissues. Insertions and substitutions in the LCAT gene may cause inactivation of the protein. Some of the reported mutations are C→T transitions on codon 147 of exon 4 (W147R), G→A on codon 293 of exon 6 (M293I), and the insertion of 3 pair of bases on exon 4, introducing a glycine on a helicoidally region of the protein and a substitution N228K24,25. The best characterized mutation is a C→T transition that results in a substitution of threonine for isoleucine in codon 123 (T123I) of the protein, resulting in a partial deficiency of LCAT29,30.
The following are some genetic alterations associated with hypercholesterolemia and hypertriglyceridemia, including their relationship with increased cardiovascular risk:
1. LDLR gene – LDL-c receptor and familial hypercholesterolemia. LDL-c is a macromolecular complex that transports cholesterol and cholesteryl esters from the liver to other peripheral tissues, where cholesterol is introduced to the cells through LDL receptors (LDLR). LDL binds to its receptors before internalization by endocytosis31. This transport represents the principal mechanism that regulates cholesterol concentration in plasma. Any defect in this transport results in hypercholesterolemia. Mutations in the LDLR gene that codifies the LDL-c receptor is one the best characterized genetic defects causing dyslipidemia. This autosomal dominant condition is called familial hypercholesterolemia32,33. Familial hypercholesterolemia is characterized by elevated levels of cholesterol and LDL-c as a consequence of defects in cholesterol transportation, receptor deficits, or a functional alteration of cellular receptors.
2. APO B-100 gene – ligand of LDL-c receptor and Familial Apolipoprotein B dysfunction (hypercholesterolemia type B). An inadequate cholesterol transport also caused by genetic defects in the ligand of LDLR, the APO B-100. This autosomal dominant defect, also known as familial APO B-100 dysfunction, comes from mutations in APO B-100 gene, in the short arm of chromosome 234. The first mutation described is a G→A transition that results in a substitution of Arg3500→Gln on the APO B-100 receptor of LDL-c33,35. Similarly, there are mutations associated with hypercholesterolemia and elevated TG that correlate with elevated CVD risk. Mutations of the LDL-r gene is the cause of familial hypercholesterolemia, which causes elevation of both total and LDL-c cholesterol27,28, and APO B-100 mutations located in p2 (transition G→A is the best known), resulting in a substitution of Arg3500→G1n in the region of APO B-100 that binds to LDL-c29, leading to type B hypercholesterolemia35.
3. APO E gene – Apolipoprotein E and hyperlipoproteinemia or hyperlipidemia type III. Apolipoprotein E (ApoE) is a principal component of chylomicrons (CMs), very low density lipoprotein (VLDL) and some HDL-c. Its main function is the hepatic clearance of CMs and VLDL, as well as lipolysis by the same lipoprotein lipase (LPL)36. In hyperlipoproteinemia or hyperlipidemia type III, plasma levels of cholesterol and TG increases as a consequence of defective transport of CMs and VLDL, due to a defect in the ApoE gene located on the short arm of chromosome 19 (19q13.2)37,38. Polymorphisms in the codifying gene of ApoE (alleles ε2, 3 y 4)39,40 are associated with variations in plasma levels of cholesterol, where the individuals with ε2 allele have 10% lower cholesterol levels compared to those who express the ε4 allele, leading to cholesterol values 10% above the mean of homozygous individuals for ε3., leads to hyperlipoproteinemia or type III hyperlipidemia with elevated total cholesterol and elevated TG41,42.
4. Lp(a) gene - lipoprotein (a)– Lp(a). Lipoprotein(a) is composed by a common low density lipoprotein (LDL-col) nuclei linked to an apolipoprotein (a) [Apo(a)] by disulfide bonds between a cysteine in the Kringle-IV type 9 (Cys 67) and the cysteine 3734 in Apo B-10043. Structurally, Apo(a) is composed of heavily glycosylated tridimensional structures called “Kringle” because of their similarity with a looped Danish pastry. Each Kringle contains a mean of 80 amino acids stabilized by 3 internal disulfide bonds, which finally surround the LDL molecules43. Apo(a) has high structural similarity with plasminogen, a key proenzyme of the fibrinolytic pathway44. Kringle IV domains are classified into 10 distinct subclasses, which compose most of the Apo(a) molecule, plus a linked Kringle V domain that resembles the catalytic region of plasminogen. The Kringle IV type 2 domain gene can be expressed a different number of times, resulting in a variable copy number of this structure (3–40 copies) within the Lp(a) molecule44. This determines the basis for the isoform size heterogeneity of Apo(a), whereas, the remaining 9 subtypes of Kringle IV are expressed just in a single copy into the Apo(a) molecule. Lp(a) is one of the most important cardiovascular risk markers45,46 and to date, some polymorphisms in the Apo(a) gene, located on chromosome 6 (6q26-q27) have been identified. For example, KIV-2 CNV consists of variable numbers of repeated units of module 4 and the number of repetitions inversely correlates with plasma levels of Lp(a)47.
5. HL gene - hepatic lipase and phenotype of combined familial hyperlipidemia. Combined familial hyperlipidemia is a genetic lipid disorder that accounts for 10–20% of premature CAD worldwide. Affected individuals’ exhibit hypercholesterolemia and/or hypertriglyceridemia and elevated concentrations of APO B, with low values of HDL-c. These are collectively called iatrogenic lipoproteinemia phenotype. There have been demonstrations of alterations in common genetic loci between families of both combined familial hyperlipidemia phenotype and iatrogenic lipoproteinemia phenotype. Such loci include genes of superoxide manganese dismutase, transport proteins of cholesteryl esters/lecithin, cholesterol acyl transferase and AI-CIII-AIV, as well as a great variety of studies relating polymorphisms in the promoter region of the LH gene (C-480T and C-514T polymorphisms) with lowering on plasma levels of HDL-c48,49.
6. LPL gene - lipoprotein lipase, Apo CII and familial dyslipidemia type O or familial chylomicronemia. Any mutations on the LPL gene, which results in a partial deficiency of the enzyme, will cause an increase in TG concentration. This is the basis of familial chylomicronemia, familial dyslipidemia type I or familial hypertriglyceridemia50. These are monogenic diseases with autosomal recessive inheritance, consisting with pure hypertriglyceridemia, TG values of 300 to 800 mg/dl, cholesterol <240 mg/dl, increases in VLDL and CMs, and lowering of LDL-c and HDL-c. To date, some LPL variants have been characterized because of amino acids substitutions in different positions (D9N, N291S, substitutions of glycine for glutamine on codon 188 and serine for a termination signal on codon 447)51. The enzymatic activity of LPL is also lowerd by mutations on the ApoC2 gene, located on chromosome 19q and codifies an essential activator of LPL52.
This information justifies the use of genetic markers for early diagnosis and cardiovascular risk assessment, especially in children and adolescents, in order to adopt early nutritional or pharmacologic interventions with the aim to mitigate atherosclerotic artery disease.
The LPL gene is located on the short arm of chromosome 8, on the region 21.3 (8p21.3). It is formed of 10 exons and 9 introns (Figure 1), and the gene codifies a protein of 475 amino acids53,54.
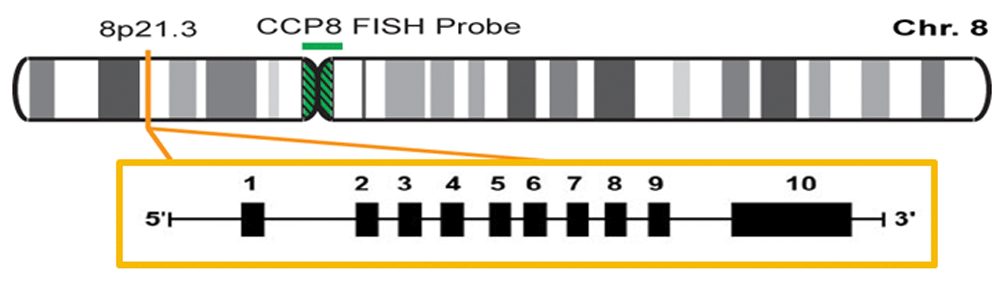
The authors confirm that this is an original image and has not been re-used or adapted from another source.
LPL is a multifunctional glycoprotein enzyme that plays an important role on lipid metabolism. After being secreted, it adheres to the luminal surface of endothelial cells where it hydrolyzes TG in circulating lipoproteins. This constitutes the limiting step on lipoprotein elimination, such as CMs from exogenous sources, and those endogenous sources, like VLDL, in circulation55,56.
In this way, LPL affects serum levels of TG, generating lipoprotein remnants that are processed by hepatic lipase. Recently, it has been demonstrated that LPL serves as a ligand for the protein related to the LDLR and influences hepatic secretion and VLDL and LDL-c capture57. Additionally, LPL has been linked to the retention of LDL-c by the sub-endothelial matrix and arterial wall, increasing LDL and VLDL conversion into more atherogenic forms58. Genetic modifications can affect LPL activity, which results in changes in lipid metabolism. Examples are slow hydrolysis of CMs and VLDL-c, longer LDL-c half-life, and decreased production of HDL59,60.
Around 100 mutations have been described on the LPL gene. The most frequent are Asp9sn, Gly188Glu and Asn291Ser. The mutations in the homozygous form are associated with hyperlipoproteinemia type I (familial chylomicronemia). Heterozygous mutations have a significant incidence in the general population (3–7%) and leads to up to a 50% decreased activity of LPL, causing an increase in TG and a decrease in HDL-c. All these lipid profile patterns increase the risk of CVD61.
Genetic studies have revealed around 100 mutations and polymorphisms in simple nucleotides on the LPL gene, some are protective, which others are deleterious:
1. Ser447x (rs328) polymorphism is located in exon 9, where cytosine is substituted by guanine on position 1959. This polymorphism leads to the suppression of both final amino acids, serine and glycine on position 447 of the protein that codifies a LPL protein prematurely truncated, which has increased lipolytic activity and increased levels of post-heparin LPL activity in X447 carriers. This is associated with the variant Ser447X, with low levels of TG, small increases of HLD-c levels, and a moderate CVD risk reduction62.
2. Pvull (rs285) polymorphism, located on intron 6, is located 1.57 kb from the Splicing Acceptor (SA) site. This polymorphism is the product of a change of cytosine for thymine. The region that contains the Pvull site is similar to the site of splicing, which interferes with the correct splicing of mRNA. However, the physiological role of this polymorphism is not completely clear yet, since it does not alter the serum concentration of lipids, nor the amino acid sequence, and a previous meta-analysis suggests that cardiovascular risk is not influenced by this polymorphism63.
3. HindIII (rs320) polymorphism is one of the most common polymorphisms of LPL gene (see below).
HindIII is a transition of intronic bases of thymine (T) to guanine (G) on position 495 of intron 8 of the LPL gene, which eliminates the restriction site for the HindIII enzyme (Figure 2 and Figure 3).
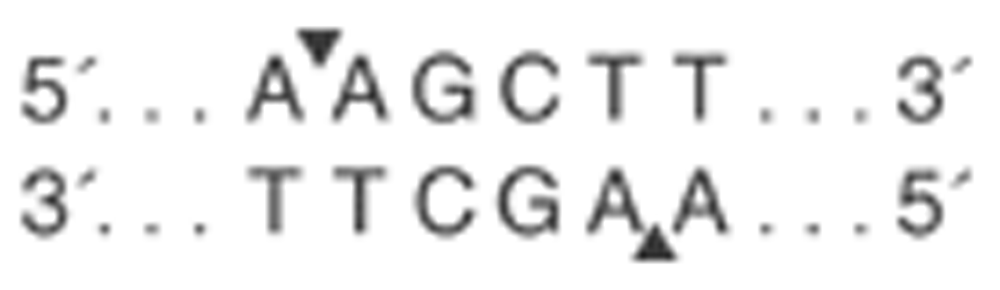
The authors confirm that this is an original image and has not been re-used or adapted from another source.

The authors confirm that this is an original image and has not been re-used or adapted from another source.
HindIII is one the most frequent polymorphisms found in various studies, which show that the homozygous genotype T/T (H+ /H+ ) represents from 45.1 to 56.4% of Iranian and south Indian populations, respectively most frequent, followed by the heterozygous T/G with 35.8–36.6% and homozygous G/G (H-/H-), with 6.93–19%64,65. Similar results have been reported in Europe66,67 and Brazil68.
The allele H+ (presence of thymine “T” or restriction site of HindIII enzyme) results in a cut on the base pair sequence in two bands of 217pb and 139pb. This is associated with a decrease in the activity of LPL in comparison with the allele H- (presence of “G” or absence of the enzymatic restriction site or presence of HindIII polymorphism). With 137pb, in which there is no cut in the LPL gene intron 8 sequence, maintaining a unique sequence of 356pb (Figure 4)69, leading to both alterations in lipidic metabolism and cardiovascular risk profile modifications in these populations.
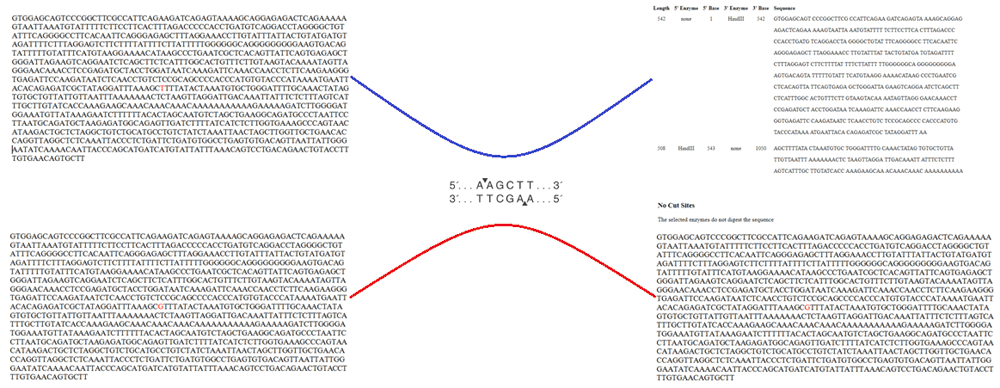
The authors confirm that this is an original image and has not been re-used or adapted from another source.
Some studies have demonstrated that the common allele (T or H+) is associated with lower levels of HDL-c in contrast with the uncommon allele (G or H-)70,71. In addition, those individuals with H+/H+ genotype had a higher concentration of serum levels of TG when compared with homozygous genotype H-/H-66,67,70,72. Similarly, there have been reports of high serum levels of LDL-c71 and a higher global cardiovascular risk in patients who carry the common allele (T or H+), see Table 1. Some studies had reported a significant drop in the LPL activity among carriers of the uncommon G allele when compared with the more common allele T57.
LPL expressed by macrophages and other cells contained in the vascular walls is involved in the early atherogenic process and is associated with increased atherosclerosis. Overexpression of LPL is also associated with insulin resistance and HTN by increased sodium retention, inflammation, vascular remodeling, sympathetic nervous system activation, oxidative stress and vasoconstriction73–75.
On the other hand, HTN (mostly systolic) has been shown to be associated with the polymorphism HindIII in the Mexican population in studies by Muñoz-Barrios et al.76. Similarly, the homozygous genotype for the common allele (H+) was associated with a higher risk of myocardial infarction in patients older than 90 years old in contrast with carriers of the uncommon allele (H-), associated with a lower prevalence of cardiovascular complications77. Clear associations were found between genotypes of LPL HindIII with HTN (H+/H+ with an OR: 2.13; 95% CI: 0.93-4.8)72 and smoking58. In a more recent study, it was established that the presence of homozygous genotype for the common allele (H+/H+) of the LPL gene is a risk factor for a first episode of myocardial infarction65. Conversely, studies by Imeni et al.78 in an Iranian population, showed no statistically significant associations between CAD and genotypic distributions of HindIII polymorphism.
Recent studies have shown increased risk of stroke among those with LPL gene variations, particularly in the HindIII gene79. He et al. reported a lower risk of stroke among patients with HindIII polymorphisms with allele G (G vs T; OR=0.78, CI95%=0.70-0.87, p<0.001). This pattern was observed in patients with ischemic stroke (G vs T. OR=0.84, CI95%=074-0.95, p=0.005) and hemorrhagic stroke (G vs. T; OR=0.60, CI95%=0.48-0.74, p<0.001)80.
From a neurologic point of view, there is scant data associating homozygous common genotype (H+/H+) with the development of Alzheimer’s disease of late appearance. This is founded on the LPL function in regulation cognitive function, mediated by cholesterol and Vitamin E transport to neuronal cells on the hippocampus and other brain areas64. These investigations appear to indicate that the HindIII polymorphism might exert a positive influence in human metabolism, which translates into improved cardiac and cerebrovascular function.
Dyslipidemias are independent risk factors for atherosclerotic artery disease. High TC, TAG and LDL-C, as well as decreased serum HDL-C, are frequently associated with low physical activity and poor eating habits, but there is a large number of mutations and single nucleotide polymorphism related to a specific protein dysfunction within major lipoprotein metabolism pathways like CETP, ApoA, LCAT, LDL receptor, Apo B-100 and LPL.
In this regard, the LPL gene HindIII polymorphism (rare allele H-) poses a protective function through its role in producing an improved lipid profile (low TG and LDL-c and high HDL-c). On the other hand, the presence of common allele (T or H+) is associated with pro-atherogenic dyslipidemias and raised cardiovascular risk. The uncommon allele (G or H-) with an absence of restriction HindIII enzyme exhibits a lower prevalence of at least 20% according to the current available literature.
There are no studies in Venezuela that allows us to know the true prevalence of the HindIII polymorphism, nor to corroborate the association with changes in the lipid profile or an increased risk for cardiovascular diseases, so we suggest performing a national populational genetic study in search for this lipidic disorders with the aim to has a better understanding of the cardiovascular risk factors in Latin America.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)