Keywords
DNA forensic, identification, mixture analysis, probability of random man not excluded
DNA forensic, identification, mixture analysis, probability of random man not excluded
An adjustment factor for linkage disequilibrium has been added to the Fast P(RMNE) formulas. Figures 1-3 have been revised.
High throughput sequencing (HTS) of DNA single nucleotide polymorphism (SNP) panels have significant advantages for analysis of DNA mixtures and trace DNA profiles compared to sizing STRs. Analysis of mixtures by sized STRs is limited to mixtures of two individuals within DNA ratios of 1:1 to 1:10. In contrast, SNP-based methods offer the potential to analyze complex mixtures of 15 contributors or more2. The current method of calculating the significance of a match between a SNP DNA mixture and a reference profile is the random man not excluded P(RMNE) calculation2 for forensic applications. However, performance and precision issues are being observed with current implementations of the P(RMNE) calculations2. To address the calculation artifacts and performance issues, a novel P(RMNE) calculation method is presented.
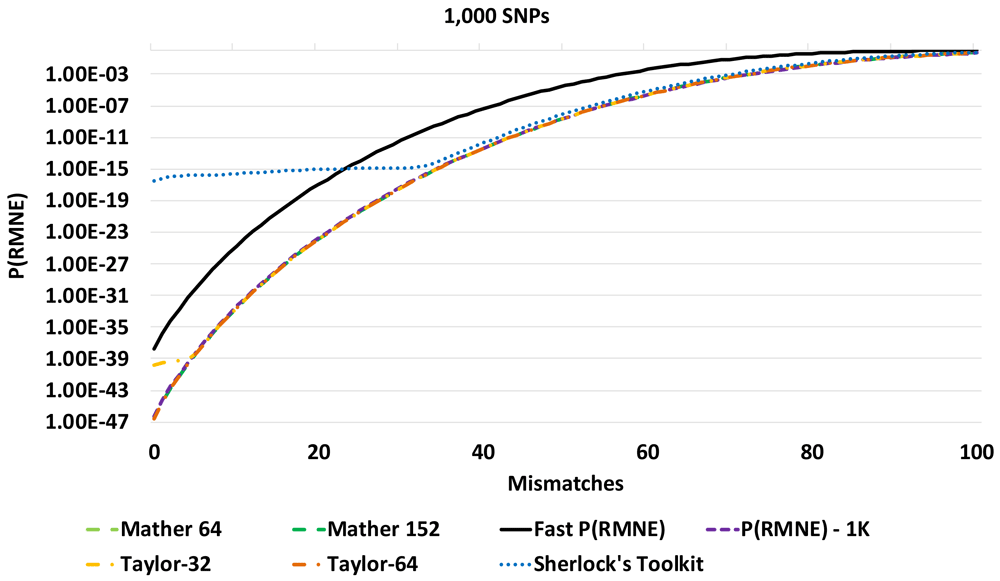
Most SNPs have just two alleles. The most common SNP allele is named the major allele. The other SNP allele(s) are named the minor allele(s). In a mixture profile, the minor allele ratio is calculated as the ratio of minor allele reads divided by the total number of reads. Methods for calculating P(RMNE) have been presented that focus on the mixture SNP loci with no called minor alleles in a mixture profile (e.g., SNPs with minor allele ratios <= 0.001 threshold)2,3. The P(RMNE) method described by Isaacson et al.2 was implemented in Sherlock’s Toolkit4. This formulation enabled P(RMNE) calculations with a small number of dropped alleles for reference profiles compared to mixture profiles. For larger DNA panels, an issue with precision was observed with the Sherlock’s Toolkit implementation, see Figure 1. This method was re-implemented in Java with higher precision libraries in an effort to eliminate the calculation artifacts observed (Figure 1). The Discrete Fourier Transform-Characteristic Function (DFT-CF) method was implemented with Taylor series approximation of trigonometric functions, named Taylor-32 for 32-bit floating point and Taylor-64 for 64-bit floating point calculations.
The Taylor series library functions were replaced with functions from Mathar’s BigDecimalMath class (http://www.mpia.de/~mathar/progs/jdocs/org/nevec/rjm/BigDecimalMath.html) to address issues detected with the Taylor-32 and Taylor-64 methods using both 64-bit and 152-bit precision.
Adjacent SNPs may be in linkage disequilibrium such that the alleles of the SNPs have non-random association with each other. The linkage disequilibrium between two SNP alleles is measured by D’ with values between 0 (unlinked) to 1 (fully linked). An adjustment factor for linkage disequilibrium is applied for SNPs ordered by chromosome position. Equation (1) represents the sum of linkage disequilibrium (LD) for the N SNPs with major alleles in a mixture. Adjusting the count of mixture SNPs with major alleles (N) by (E) approximates the number of unlinked SNPs with major alleles in a mixture.
An alternative to the DFT-CF P(RMNE) method was implemented. A mixture will have N loci with no called minor alleles. Let p be the average minor allele ratio at these mixture loci. Let q be defined as 1 – p such that p + q = 1. SNP panels can be optimized for DNA mixture analysis2,3; the average of the SNP minor allele ratios used for a P(RMNE) calculation can be used to approximate large numbers of individual SNPs with similar minor allele ratios. For an individual with two alleles at a SNP loci the probability for these alleles can be represented as (p+q)2 = p2 + 2pq + q2 = 1. A perfect reference match to a mixture has major:major (MM) alleles at every locus with no called minor alleles in the mixture profile. Mismatches are defined as reference loci with major:minor (mM) or minor:minor (mm) at these mixture loci with no called minor alleles (MM). The number of mismatches is defined as L between a reference and a mixture. Let K be (1 – q2)/q2 represent the ratio of transition from MM to non-MM (i.e., mM or mm). Let Combination represent the standard statistics combination operation for representing possible SNP loci that mismatch between a reference and a mixture (1). PRMNE(L) can be estimated by the term for no mismatches, q2(N-E), times the possible combinations of L mismatches, Combination(N-E, L), times the transition term KL (2)2. Equation (3) illustrates the calculation for no mismatches (L=0), and (4) for one mismatch (L=1). Consecutive terms can be calculated efficiently for multiple L values as illustrated by (5) and (6). This optimization has the additional benefit of multiplying a large value, (N-E-L)/(L+1), with a small value, K, where calculating (N-E)!/L!(N-E-L)! by itself can stress the precision capability of an implementation for large values for N-E and L. Equation (7) represents the P(RMNE) calculation for 0 to L mismatches.
Timing for the Sherlock’s Toolkit (Python), Taylor, and Mathar algorithms (Java) were run on an Intel Xeon E5-2609 v2 2.5 GHz dual CPU system with 32 GB RAM. Fast P(RMNE) (Ruby) was run on a MacBook Pro laptop with 2.8 GHz Intel i7, 16 GB 1600 MHz DDR3 RAM, 750 GB SSD hard drive.
The calculated P(RMNE) values for Sherlock’s Toolkit and Taylor-32 both have calculation artifacts/precision issues compared to the Taylor-64 method for a panel of 1,000 SNPs in Figure 1. The Sherlock’s Toolkit P(RMNE) values start to deviate from actual P(RMNE) values with 36 or less mismatches while the Taylor-32 deviates at 5 or less mismatches. When the panel size is increased to 3,000 SNPs, the Taylor methods are unable to calculate P(RMNE) values. For higher precision, the Mathar BigDecimalMath library was used with 64-bit and 152-bit precision. Calculation artifacts are seen for the Mathar 64-bit method for the 3,000 SNP panel (Figure 2) and the Mathar 152-bit method for the 4,000 SNP panel (Figure 3). The root mean square error (RMSE) between Fast P(RMNE) and Mathar-152 was 2.2e-41. This calculation excluded the Mathar 152-bit calculation artifacts between 0 and 19 mismatches. Algorithm timing results are shown in Figure 4. For the 1,000 SNP panel, the Taylor 64-bit algorithm runs in 142 s and the Taylor 152-bit in 1,017 s. The Taylor methods did not complete for the larger panel sizes.
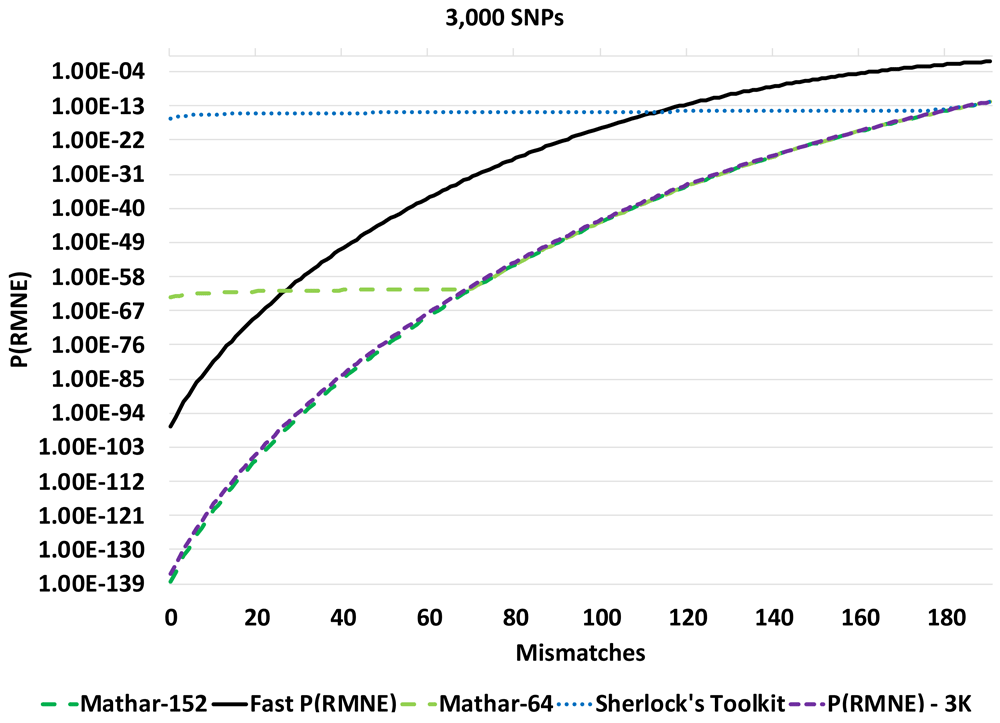
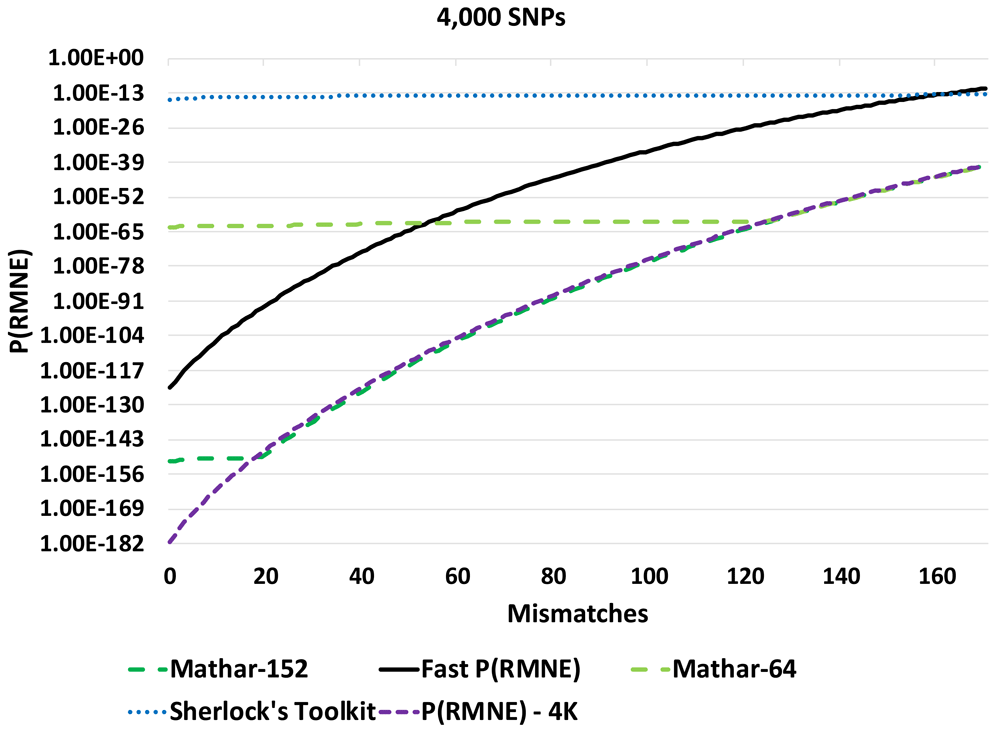
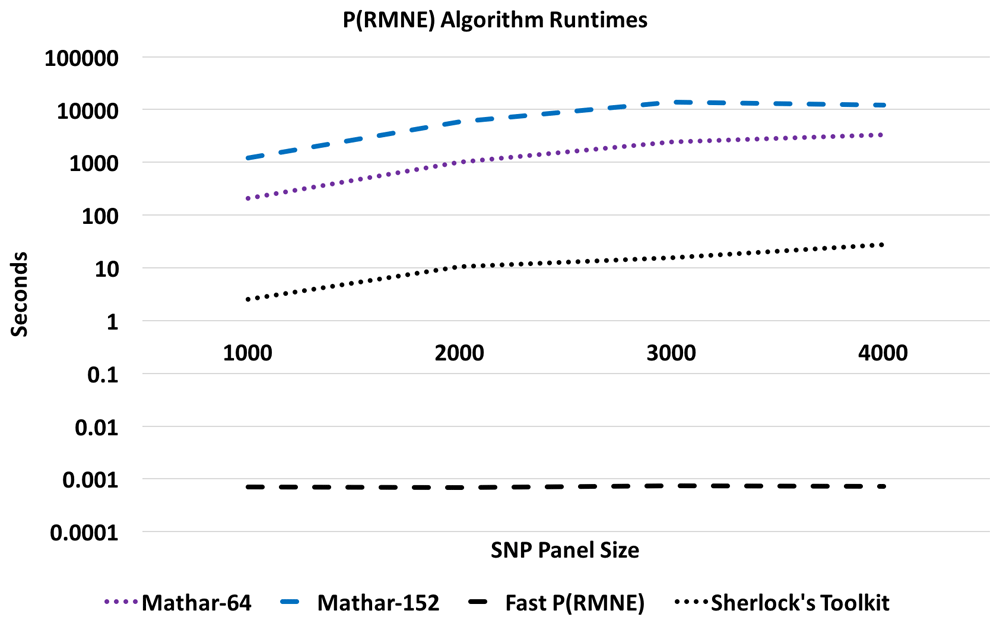
A calculation artifact was observed for some datasets with the P(RMNE) method implemented in Sherlock’s Toolkit, see Figure 1. Shifting to higher precision libraries, improved the results for smaller SNP panels, but calculation artifacts appear for larger SNP panels, see Figure 2 and Figure 3. Also, the Taylor methods crash with larger panels or return no results. The Mathar BigDecimalMath libraries work better than the Taylor method library, but calculation artifacts are again observed for the 4,000 SNP panels for both Mathar-64 and Mathar-152 methods. The runtimes for these higher precision methods as increased beyond what was desirable for rapid forensic sample analysis. The Fast P(RMNE) method addresses both the calculation artifact issue (Figure 3) and the runtime issue (Figure 4). Equation (6) enables the rapid calculation of P(RMNE) for a series of possible mismatches in a fraction of a second on any modern CPU processor. Adjusting for linkage disequilibrium in SNP panels provides an improved estimate of P(RMNE).
The calculations and SNP panel data for each method and SNP panels used are included in Ricke, Darrell, 2017, “Fast P(RMNE) Data”, doi: 10.7910/DVN/ZUN3GD, Harvard Dataverse.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)