Keywords
optogenetics, archaerhodopsin, protein structure prediction, QM/MM, spectral tuning in rhodopsins
optogenetics, archaerhodopsin, protein structure prediction, QM/MM, spectral tuning in rhodopsins
Additional information and data analysis were added in Results section of the article: 1. The comparison of results of different sequence alignment algorithms (new Figure 1 provided); 2. The comparison of the obtained model of archaerhodopsin-3 with the crystallographic structure of archaerhodopsin-1 (new Figure 2, Figure 3); 3. Analysis of the results of absorption maxima calculations for archaerhodopsin-3. Information about analogous results for other rhodopsins is provided. 4. Residues replaced in site-directed mutagenesis study (Mclsaac RS et al, PNAS, 2014) were visualized (new Figure 4).
Modifications made in the Introduction section and Abstract, considering the remarks of our referees. In the Methods section information about sequence identity between archaerhodopsin-1 and archaerhodopsin-3 is provided.
Also, the new variant of the archieve with input and output files, extra grant information (Russian Foundation for Basic Research and Presidium of the RAS) and acknowledgements to two computational research facilities (HPC computing resources at Lomonosov Moscow State University and Computer Center of SPbU) are provided.
The additional grants listed, have aided in the continuation of the project that was supported by previous funding.
See the authors' detailed response to the review by Adam E. Cohen and Samouil Farhi
See the authors' detailed response to the review by Jeremy N. Harvey
Precise and quick control of physiological processes using integrated optical and genetic methods is a vast area with a high number of important applications1. One possible approach in this field is using fluorescent voltage-dependent indicators for detecting the activity of mammalian neurons, which allows achievement of the precision of a single neuron without perceptible time delays. It was recently shown that some rhodopsins, especially achaerhodopsin-3, can be potential candidates for such a task2–5. While the results obtained for archaerhodopsin-3 are already very encouraging, increasing the fluorescent signal of such proteins can lead to a significant progress in this field. It was also shown that the insertion of mutations into these proteins can dramatically improve the signal quality2–5.
Unfortunately, the fundamental mechanisms underlying the processes that determine fluorescence are not well understood. This lack of knowledge leads to difficulties with design of desired rhodopsin mutants. While rational design is not based on a solid foundation and, for this reason, is not very effective, another experimental approach, random mutagenesis, is very time consuming. Computational studies can provide additional insights into the problem. One of the main obstacles for computational modeling of proteins is the absence of three-dimensional structures of high quality, especially for membrane proteins, which are a challenge for crystallization. On the other hand, computational prediction of three-dimensional structures is not trivial. The goal of this study was to obtain a good-quality structure for achaerhodopsin-3, one of the most used voltage-dependent fluorescent sensors4,5, and based on this structure to predict the optical properties of this protein.
To achieve this goal, we used a homology-based computational approach for structure prediction. As the choice of a structure prediction algorithm is not straightforward, we tested several methods. To evaluate the quality of obtained structures we performed subsequent Quantum Mechanics/Molecular Mechanics (QM/MM) calculations of absorption maxima and compared the results with available experimental data.
The structure of archaerhodopsin-3 was built using a homology modeling approach. Primary structures for all rhodopsins with crystallographic data are available in the Protein Data Bank library6 (24 structures as of September 2016) were compared with the primary structure of archaerhodopsin-3. Archaerhodopsin-1, which has the highest sequence identity to the target protein, was chosen as a template (RCSB code 1UAZ, sequence identity 84.5%, sequence similarity 91.5%). Three algorithms of homology-based model building were tested: Medeller7, I-TASSER8 and RosettaCM9. All methods of homology modeling heavily rely on externally made target-template alignment of primary sequences, which serves as the main instruction for model building. For this reason, we tested three algorithms of pairwise alignment using their results as an input for each method of model building. Two of the alignment methods are specifically constructed for membrane proteins, MP-T10 and AlignMe11, and the third one, MUSTER12, gains its quality from evolutionary predictions. The latter algorithm is a built-in algorithm of I-TASSER suite and its results were used only for this method of structure prediction.
Before QM/MM calculations, several preparation steps were performed: hydrogen atoms were added using pdb2pqr package13 version 2.1.1 using CHARMM force field version 2714, pH=7; hydrogen atoms were equilibrated by energy minimization in NAMD package15, version 2.11. The retinal chromophore was bound to the lysine residue Lys226, whole lysine + retinal system was parameterized in CHARMM force field16. The protein was inserted in the POPC membrane17, the whole system was inserted in a water solvent box, the TIP3P water model18 was used, the size of water box was selected so that there were at least 10 Å from any atom of protein to the edge of the system, and the system was neutralized by addition of Na+ and Cl- ions.
Relaxation of the system was performed in several steps: relaxation of retinal + lysine complex with all other atoms fixed, relaxation of all atoms that were within 6 Å of the chromophore system, relaxation of whole protein and water box. During all these steps the following parameters were used: a 10 Å cutoff with switching starting at 8.5 Å was applied to the electrostatics and van der Waals interactions; Particle Mesh Ewald method19 was used for dealing with electrostatics interactions, grid spacing 1Å. Equilibrated protein structure was extracted; internal waters were added into protein cavities using WaterDock program20.
To calculate absorption maxima, we used the methodology that has been proven as efficient in a number of our previous studies for different kind of rhodopsins and rhodopsin mimics21–27. The structures of the archaerhodopsin-3 obtained at the previous step were optimized using two-layer ONIOM (QM:MM-EE) scheme. (QM=B3LYP/6-31G*; MM= AMBER for aminoacids and TIP3P for water, EE=electronic embedding) was implemented in the Gaussian09 package28. To calculate the spectral properties of the chromophore in the presence of the protein environment (described as AMBER point charges) SORCI+Q/6-31G* level of the theory was used, as it is implemented in ORCA6.0 package29. For details of previously performed QM/MM methodology see Altun et al.30,31.
On the first step of structure prediction of archaerhodopsin-3 we performed the alignment of its amino acid sequence with the sequence of archaerhodopsin-1, comparing three different algorithms: AlignMe, MUSTER and MP-T. The results of AlignMe and MUSTER algorithms were identical; they differ from the MP-T alignment result in the flexible N-terminal part (Figure 1, a–c).
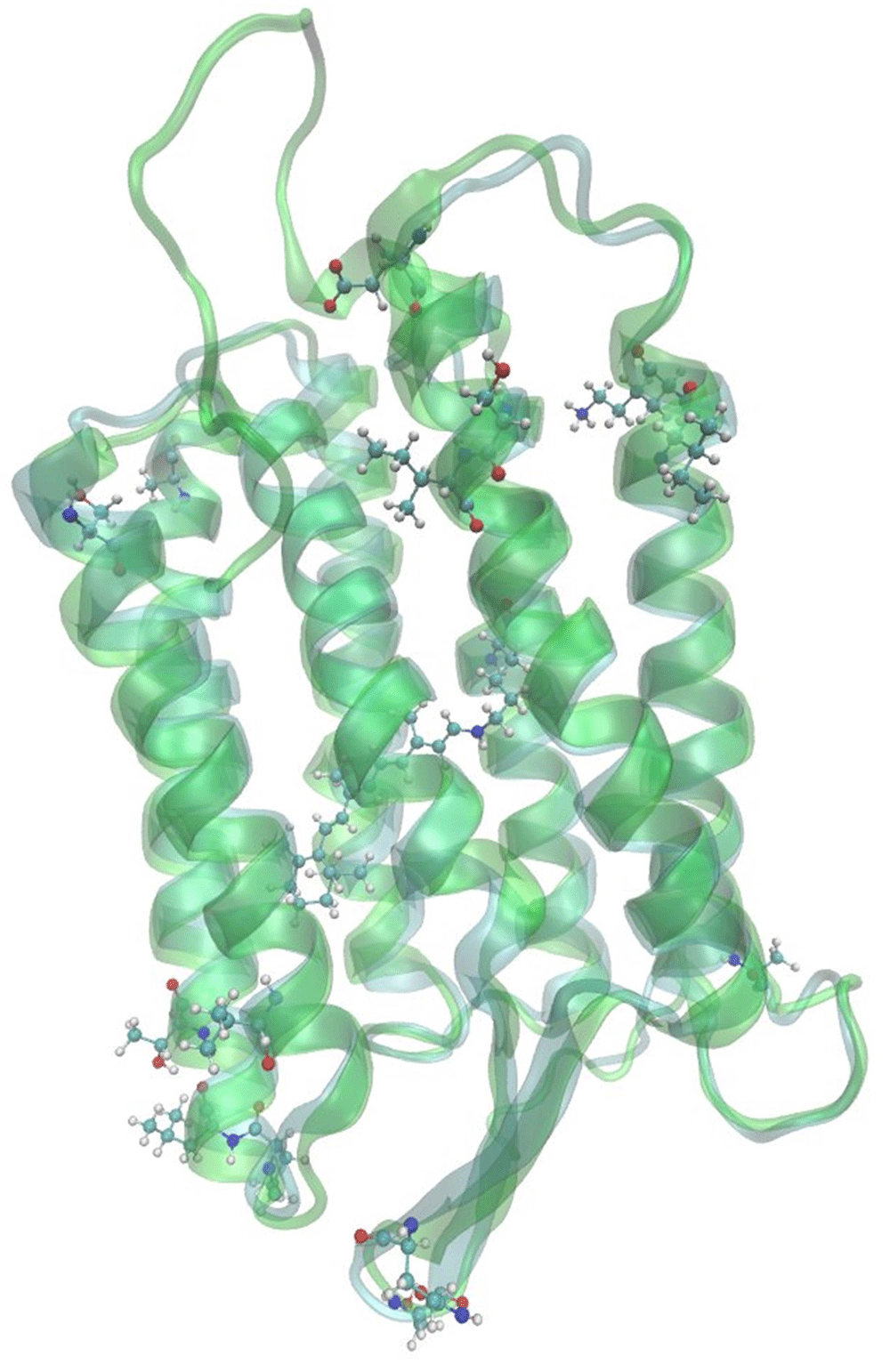
On the next step we built 7 three-dimensional models of archaerhodopsin-3 using I-TASSER, Medeller and RosettaCM algorithms of template-based structure prediction. Comparison of crystallographic structure of archaerhodopsin-1 with the predicted structure of archaerhodopsin-3 showed that these two proteins differ only in loop regions and on the edges of the alpha-helices (Figure 2, Figure 3). These regions are located on relatively large distance from the retinal chromophore. We also highlighted the residues, which were the sites for the site-directed mutagenesis of the archaerhodopsin-35 (Figure 4). The computational characterization of these mutants is an interesting problem for further investigations.
For all seven predicted models we calculated the absorption maximum wavelengths. All obtained models provide reasonable results -- the deviation range is only 31 nm from the experiment (Table 1). This deviation range is sensible considering the approximations used in our methodologies. In our previous studies, we obtained a 29 nm shift for the absorption maximum of halorhodopsin from N.pharaonis23, a 25 nm shift for the absorption maximum of archaerhodopsin-2 and a 39 nm shift for the channelrhodopsin-2. The model with the smallest deviation was predicted by I-TASSER algorithm with AlignMe alignment (Figure 2).

The results of the pairwise sequence alignment algorithms: a) AlignMe; b) MP-T; c) MUSTER.


In this study, we predicted the structure of fluorescent voltage-dependent sensor achaerhodopsin-3 and evaluated its quality with subsequent QM/MM high level ab initio calculations of spectral properties. The calculated absorption maximum is within 31 nm from the experimental value. Several methods of model building were tested and spectral characteristics were calculated for all resulting models. We showed that our methodology allowed for reliable prediction of optical properties of archaerhodopsin-3. The results of this study can be utilized for high-level QM/MM investigation of different aspects of photochemistry of this voltage-dependent fluorescent sensor and, therefore, to contribute in development of the efficient molecular tools for modern optogenetics.
The sequence of archaerhodopsin-3 was taken from Uniprot database: http://www.uniprot.org/uniprot/P96787
The template for homology modeling was taken from PDB database (rcsb code 1UAZ): http://www.rcsb.org/pdb/explore/explore.do?structureId=1UAZ
The input files of I-TASSER suite, RosettaCM, Medeller algorithms with corresponding README files, zipped output of I-TASSER suite, scripts for processing structure after homology modeling stage (with instructions in README file), input files for spectra calculations are available: doi, https://doi.org/10.5281/zenodo.83002532.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)