Keywords
Diabetes, neuropathic, pain, neurosteroid, benzodiazepine, ob/ob, mitochondria, TSPO, GABA
Diabetes, neuropathic, pain, neurosteroid, benzodiazepine, ob/ob, mitochondria, TSPO, GABA
Changes have been made in response to comments made by the reviewers. The title has been made more specific to the data presented. In the Abstract, "neuropathic pain" has been changed to "neuropathy" as the concept of pain is specific to humans, in contrast to nociception, which applies to mice and other species including humans. The Discussion has been edited to facilitate the reader’s understanding by putting the data in the context of recent work (Humble, 2016a). Similarly, Figure 1 and its legend have been edited in order to highlight action sites of diazepam, flumazenil and finasteride and therefore enhance readability. We have removed the mention of Dr Ladas from the Acknowledgements section, as this was accidentally added to the version 1.
See the author's detailed response to the review by Pascal Darbon
See the author's detailed response to the review by Slobodan M. Todorovic
Diabetic neuropathy is a common cause of painful neuropathy, and treatment is often suboptimal because the underlying aetiology is poorly understood. Peripheral and central sensitisation are implicated in the development of neuropathic pain with neuroplasticity occurring at multiple levels of the pain pathway (Harvey & Dickenson, 2008). GABAergic neurones at all levels of the pain pathway have a vital role in the transmission of painful stimuli in the perception of pain itself (D’Mello & Dickenson, 2008). Endogenous and exogenous neurosteroids may act as potent positive allosteric modulators of GABAA receptors (GABAARs) and consequently exhibit analgesic, anxiolytic, anticonvulsant, and sedative properties (D'Hulst et al., 2009).
Within inhibitory synapses, the presynaptic fusion of a single vesicle releases the inhibitory neurotransmitter GABA to activate synaptic GABAARs. Under voltage-clamp conditions this causes a miniature inhibitory postsynaptic current (mIPSC). Drugs that enhance GABAAR function cause a prolongation of the mIPSC decay phase. Recent work in a model of type-II diabetic neuropathy (ob/ob) revealed that layer 2/3 cortical pyramidal neurones of the pain pathway exhibited reduced endogenous neurosteroid modulation of the GABAAR, and exogenously applied neurosteroids had an exaggerated impact (Humble, 2016a). The mechanism responsible appeared unrelated to GABAAR sensitivity, but instead was associated with a reduction of neurosteroid precursors, such as pregnenolone, which is metabolised sequentially to the active compound allopregnanolone (Figure 1) (Humble, 2013; Humble, 2016a). Pregnenolone is synthesised in the mitochondrion from its precursor cholesterol by the side chain cleavage enzyme P450 located in the inner mitochondrial membrane (Do Rego et al., 2009; Schumacher et al., 2012), and it is postulated that diabetic neuropathy may be associated with a reduction in mitochondrial activity. Cholesterol is translocated across the mitochondrial membrane by the 18 kDa translocator protein (TSPO) in a coordinated fashion with the steroidogenic acute regulatory (StAR) protein (Do Rego et al., 2009; Gatliff & Campanella, 2016; Rupprecht et al., 2010; Stocco et al., 2017; Figure 1).
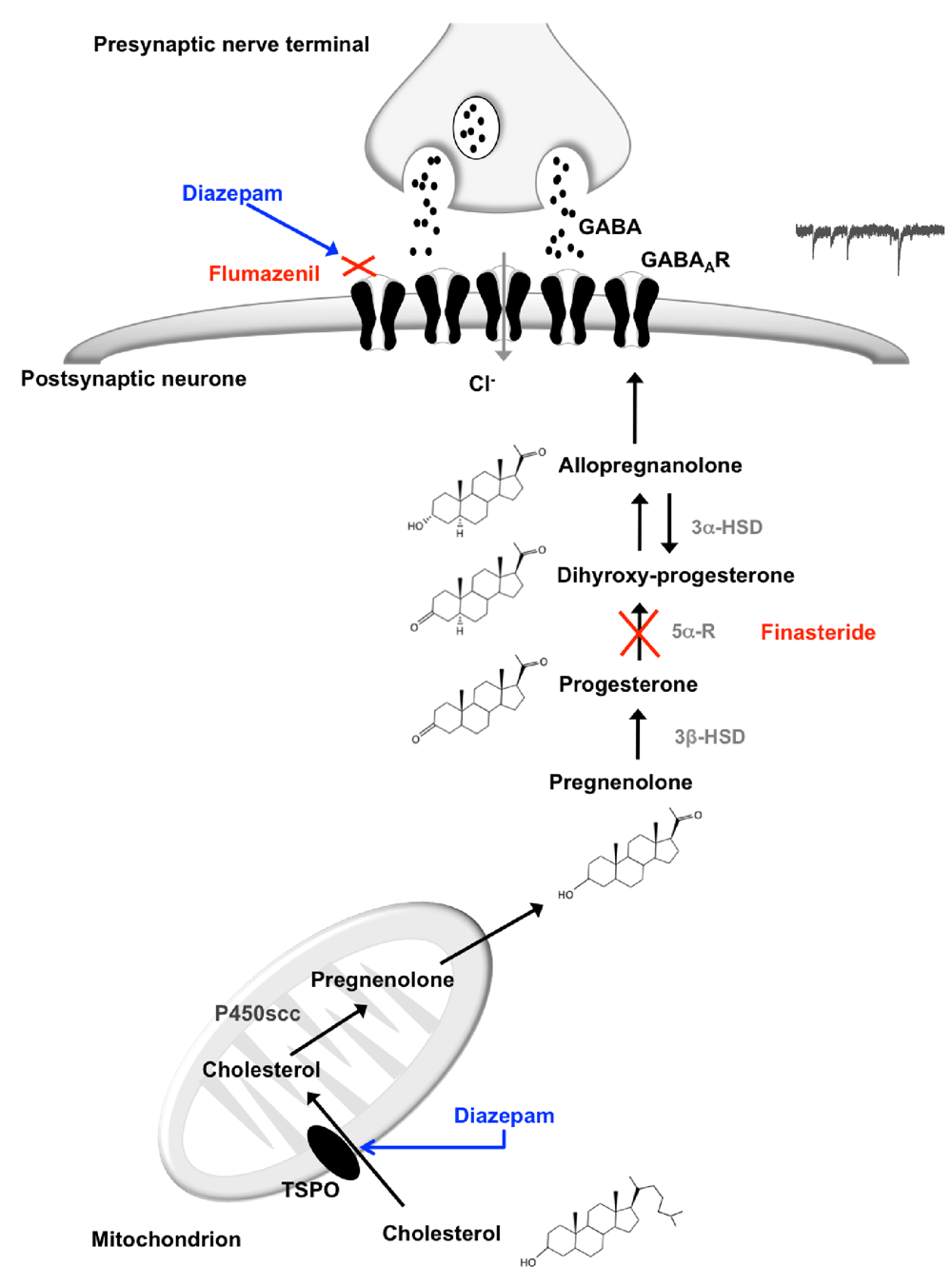
Cholesterol is taken through the mitochondrial membrane by the translocator protein (TSPO) where it is converted to pregnenolone by the cytochrome P450 side chain cleavage enzyme. Pregnenolone is converted to progesterone by 3β-hydroxysteroid dehydrogenase (3β-HSD), which is in turn reduced to dihydroxyprogesterone by 5α-reductase (5α-R). Dihydroxyprogesterone is converted to allopregnanolone by 3α-hydroxysteroid dehydrogenase (3α-HSD). Postsynaptic GABAARs are activated by GABA that has been released from vesicles in the presynaptic nerve terminal. GABA induces a conformational change of the GABAAR, opening its central channel and thereby allowing the passage of chloride ions and the subsequent generation of miniature inhibitory postsynaptic currents (mIPSCs). The negative chloride ions induce hyperpolarisation of the neuronal membrane, which mediates neuronal inhibition. Neurosteroids, such as the active compound allopregnanolone, modulate GABAAR function and facilitate inhibition of the neuronal membrane. (Humble, 2013). The translocation of cholesterol into the mitochondria by TSPO is the first rate-limiting step and is enhanced by the presence of specific ligands such as diazepam. Thus diazepam may enhance GABAAR modulation by binding to the GABAAR directly and separately by increased neurosteroidogenesis. This modulation may be selectively inhibited at the GABAAR itself by the antagonist flumazenil and separately by the 5α-R inhibitor finasteride, which inhibits neurosteroidogenisis.
The present study explored the impact of the benzodiazepine diazepam, a positive allosteric modulator of the GABAAR (D’Hulst et al., 2009), on GABAAR modulation via neurosteroidogenesis in diabetic and wild type (WT) mice. Benzodiazepines are also known to activate neurosteroidogenesis by binding to TSPO (Rupprecht et al., 2010; Tokuda et al., 2010).
The methods are identical to those published by the same author previously (Humble, 2016a), with the exception of the drugs diazepam and flumazenil, which were not used in the previous study. Diazepam and flumazenil were purchased (Tocris, Bristol UK) and prepared as concentrated stock solutions in dimethyl sulfoxide before being added to the artificial extracellular solution as per the previous study (Humble, 2016a).
Whole-cell voltage-clamp recordings were made in L2/3 cortical neurones of WT and ob/ob mice after at least two hours of incubation with diazepam, flumazenil and finasteride. Diazepam alone had the same effect on both strains of mice. In the WT mice, flumazenil inhibited the effect of diazepam (τW: control = 4.0 ± 0.1 ms, n = 35; finasteride 50 μM = 4.2 ± 0.1 ms, n = 7; diazepam 1 μM = 5.9 ± 0.2 ms, n = 6; flumazenil 10 μM & diazepam 1 μM = 4.0 ± 0.2 ms, n = 7; flumazenil 10 μM, finasteride 50 μM & diazepam 1 μM = 4.0 ± 0.3 ms, n = 6; One-way ANOVA, P <0.05; post hoc Newman Keul’s test revealed a difference only for diazepam 1 μM, P <0.05; Figure 2). By contrast, in the ob/ob mice, flumazenil only partially inhibited the effect of diazepam, and the persisting effect of diazepam in the presence of flumazenil in the ob/ob mice could be prevented by the presence of the 5α-reductase enzyme inhibitor finasteride (τW: ob/ob control = 3.5 ± 0.1 ms, n = 25; finasteride 50 μM = 3.7 ± 0.2 ms, n = 6; diazepam 1 μM = 5.7 ± 0.3 ms, n = 6; flumazenil 10 μM & diazepam 1 μM = 4.9 ± 0.3 ms, n = 6; flumazenil 10 μM, finasteride 50 μM & diazepam 1 μM = 3.7 ± 0.1 ms, n = 5; One-way ANOVA, P <0.05; post hoc Newman Keul’s test revealed significant intergroup differences for the flumazenil groups, P <0.05; Figure 2).
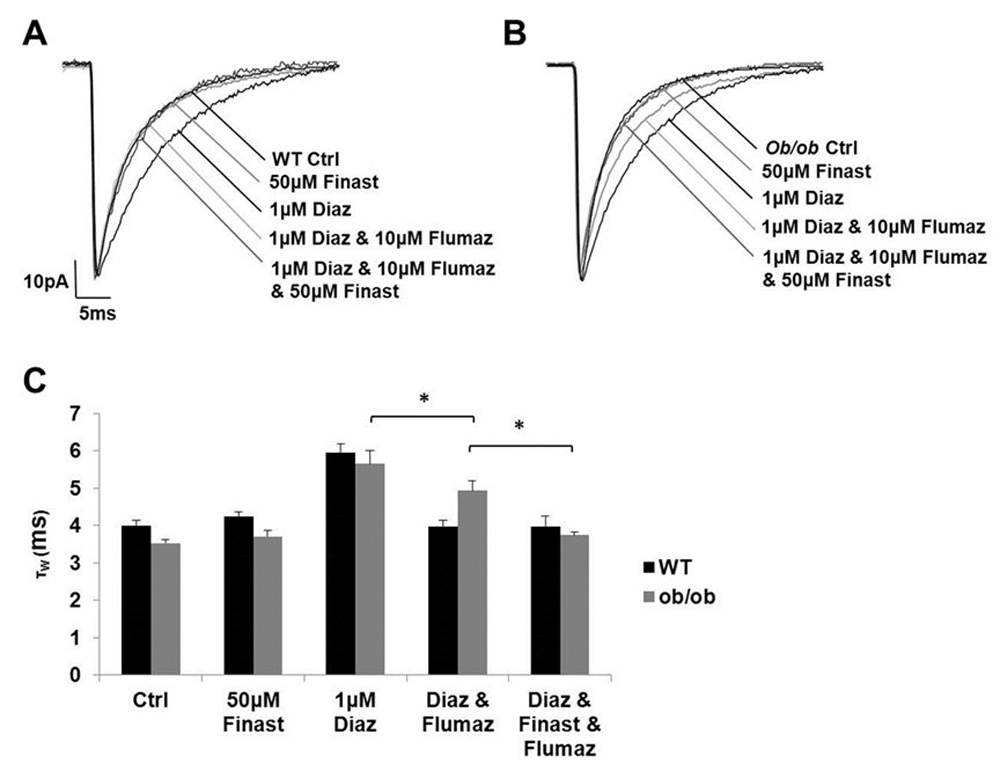
(A) Superimposed exemplar averaged GABAAR-mediated miniature inhibitory postsynaptic currents (mIPSCs) acquired from a representative WT cortical neurone and from equivalent neurones after ~2 hours pre-incubation of the brain slice with diazepam (1 μM), flumazenil (10 μM) and finasteride (50 μM). (B) Superimposed exemplar averaged GABAAR-mediated mIPSCs acquired from a representative ob/ob cortical neurone and from equivalent neurones after ~2 hours pre-incubation of the brain slice with diazepam (1μM), flumazenil (10 μM) and finasteride (50 μM). (C) Histogram illustrating that flumazenil is able to prevent the effect of diazepam to prolong the duration of the GABAAR-mediated mIPSC in WT cortical neurones, but only has a partial efficacy in ob/ob cortical neurones (τw in ms; one-way ANOVA, P >0.05; Post hoc Newman Keul’s test). The persisting effect of diazepam in the presence of flumazenil in the ob/ob mice could be prevented by the presence of the 5α-reductase enzyme inhibitor finasteride (τw in ms; one-way ANOVA, P >0.05; Post hoc Newman Keul’s test). Ctrl = control; Finast = finasteride; Diaz = diazepam; Flumaz = flumazenil.
Layer 2/3 cortical neurones from mature type-II diabetic ob/ob are known to have a reduced endogenous pregnane-derived neurosteroid tone in comparison to strain matched WT controls (Humble, 2016a). The present data indicate that by promoting the uptake of pregnenolone’s precursor cholesterol by the mitochondria, via TSPO, diazepam may rescue the reduced neurosteroid tone. The restored neurosteroid tone could re-establish GABAAR-mediated neuro-inhibitory tone in cases of neuropathic hypersensitivity. With specific reference to these data, the key result is the difference in response of the WT and ob/ob to simultaneous incubation with diazepam and flumazenil. In contrast, diazepam and the 5α-reductase inhibitor finasteride individually or in combination produced the same response in both WT and ob/ob. This may be interpreted as follows: in the WT, the primary effect of diazepam incubation is direct allosteric modulation of the GABAAR, with negligible contribution from neurosteroidogenesis via mitochondrial TSPO activation. In comparison, diazepam has an exaggerated effect on GABAergic inhibitory tone in the ob/ob, despite the presence of the GABAAR benzodiazepine antagonist flumazenil. This effect is likely observed due to physiological upregulation of the key rate-limiting enzymes involved in neurosteroidogenesis in response to the reduced pregnenolone synthesis by the mitochondria (Figure 1; Humble, 2016a). Thus by increasing the availability of the neurosteroid precursor pregnenolone via TSPO activation, it is possible to promote enhanced neurosteroidogenesis and thereby increase GABAergic inhibitory tone via an alternate route. Benzodiazepines modulate the GABAAR by binding to the α-γ subunit interface (D'Hulst et al., 2009), while neurosteroids bind the GABAAR from a cavity within the α-subunit domain and modulate it directly via the α-β subunit interface (Hosie et al., 2006). There have already been a number of other studies of ligands for the mitochondrial TSPO, as this is a promising target (Gatliff & Campanella, 2016; Giatti et al., 2009; Papadopoulos & Lecanu, 2009; Rupprecht et al., 2010; Zhang et al., 2016). Considering the findings in this paper alongside previous work (Humble, 2016a) it appears that mitochondrial dysfunction may play an important role in the development of type 2 diabetic neuropathy. In this context, it follows that enhancing the GABAergic neurosteroid tone directly or indirectly could be of potential therapeutic benefit for diabetic neuropathic pain and hypersensitivity.
Open Science Framework: Dataset of ‘Neurosteroids are reduced in diabetic neuropathy and may be associated with the development of neuropathic pain’, doi: 10.17605/osf.io/bk3tw (Humble, 2016b). Raw data for the present study can be found in Diazepam.zip.
Please refer to (Humble, 2016a) for details of standard software used for data analysis.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)