Keywords
transcriptome, sequencing, RNAseq, automation, cloud computing,
This article is included in the Container Virtualization in Bioinformatics collection.
transcriptome, sequencing, RNAseq, automation, cloud computing,
High-performance computing based bioinformatic workflows have three main subfamilies: in-house computational packages, virtual-machines (VMs), and cloud based computational environments. The in-house approaches are substantially less expensive when raw hardware is in constant use and dedicated support is available, but internal dependencies can limit reproducibility of computational experiments. Specifically, “superuser’” access needed to deploy container-based, succinct code encapsulations (often referred to as "microservices" elsewhere) can run afoul of normal permissions, and the maintenance of broadly usable sets of libraries across nodes for users can lead to shared code dynamically linking to different libraries under various user environments. By contrast, modern cloud-based approaches and parallel computing are forced by necessity to offer a user-friendly platform with high availability to the broadest audience. Platform-as-a-service approaches take this one step further, offering controlled deployment and fault tolerance across potentially unreliable instances provided by third parties such as Amazon Web Service Elastic Compute Cloud (AWS EC2) and enforcing a standard for encapsulation of developers' services such as Docker. Within this framework, the user or developer cedes some control of the platform and interface, in exchange for the platform provider handling the details of workflow distribution and execution. This has provided the best compromise of usability and reproducibility when dealing with general audiences. In this regard, the lightweight-container approach exemplified by Docker lead to rapid development and deployment compared to VMs. Combined with versioning of deployments, it is feasible for users to reconstruct results from an earlier point in time, while simultaneously re-evaluating the generated data under state-of-the-art implementations.
Several recent high impact publications used cloud-computing work flows such as CloudBio-linux, CloudMap1 and Mercury2 AWS EC23. The CloudBio-linux software is centered around comparative genomics, phylogenomics, transcriptomics, proteomics, and evolutionary genomics studies using Perl scripts3. Although offered with limited scalability, the CloudMap software allows scientists to detect genetic variations over a field of virtual machines operating in parallel3. For comparative genomic analysis, the Mercury workflow2 can be deployed within Amazon EC2 through instantiated virtual machines but is limited to BWA and produces a variant call file (VCF) without considerations of pathway analysis or comparative gene set enrichment analyses. The effectiveness for conducting genomic research is greatly influenced by the choice of computational environment. The majority of RNAseq analysis pipelines consist of read preparation steps, followed by computationally expensive alignment against a reference. Software for calculating transcript abundance and assembly can surpass 30 hours of computational time4. If known or putative transcripts of defined sequences are the primary interest, then pseudoalignment, which is defined as near-optimal RNAseq transcript quantification, is achievable in minutes on a standard laptop using Kallisto software4. After verifying these numbers on our own laptops, we became interested in a massively parallel yet easy-to-use approach that would allow us to perform the same task on arbitrary datasets, and reliably interpret the output. In collaboration with Illumina (San Diego, USA) we found that the available BaseSpace platform was already well-suited for this purpose, with automated ingestion of the Sequence Read Archive (SRA) datasets as well as newly produced data from core facilities using recent Illumina sequencers. The design of our framework emphasizes loose coupling of components and tight coupling of reference transcriptome annotations; nonetheless, the ease of use and massive parallelization provided by BaseSpace offers excellent default execution environment.
The BaseSpace Platform utilizes AWS cc2 8x-large instances by default, each with access to eight 64-bit CPU cores and virtual storage of over 3 terabytes. Published BaseSpace applications, which undergo rigorous review by Illumina staff scientists before deployment, can allocate up to 100 such nodes, distributing analyses simultaneously, in parallel. Direct imports of existing experiments from SRA, along with default availability of experimenters' own reads, fosters a critical environment for independent replication and reanalysis of published data.
A second bottleneck in bioinformatic workflows, hinted at above, arises from the frequent transfer and copying of source data across local networks and/or the Internet. With a standardized deployment platform, it becomes easier to move executable code to the environment of the target data, rather than transferring massive datasets into the environment where the executable workflows were developed. For instance, an experiment from SRA with reads totaling 141.3GB is reduced to summary quantifications totaling 1.63GB (nearly two orders of magnitude) and a report of less than 10MB (a further two orders of magnitude), for a total reduction in size exceeding 4 orders of magnitude with little or no loss of user-visible information. Moreover, the untouched original data is never discarded unless the user explicitly demands it, something that can rarely be said of local computer environments. Moreover, the location of original sources is always traceable. The appropriate placement of Arkas cloud computational applications in close proximity to the origin of sequencing data removes cumbersome data relocation costs.
The scale and complexity of sequencing data in molecular biology has exploded in the 15 years following completion of the Human Genome Project5. Furthermore, as a dizzying array of sequencing protocols have been developed to open new avenues of investigation, a much broader cross-section of biologists, physicians, and computer scientists have come to work with biological sequence data. The nature of gene regulation (or, perhaps more appropriately, transcription regulation), along with its relevance to development and disease, has undergone massive shifts propelled by novel approaches, such as the discovery of evolutionarily conserved non-coding RNA by enrichment analysis of DNA and isoform-dependent switching of protein interactions6. What sometimes gets lost within this excitement, however, is the reality that biological interpretation of these results can be highly dependent upon both their extraction and annotation. A rapid, memory-efficient approach to estimate abundance of both known and putative transcripts substantially broadens the scope of experiments feasible for a non-specialized laboratory. Recent work on the Kallisto pseudoaligner4, amongst other k-mer based approaches, has resulted in such an approach.
In order to leverage these recent advances for large scale needs, we created a cloud computational pipeline, Arkas, which encapsulates Kallisto, automates the construction of composite transcriptomes from multiple sources, quantifies transcript abundances, and implements reproducible rapid differential expression analysis followed by a gene set enrichment analysis over Illumina's BaseSpace Platform. The Arkas workflow is versionized into Docker containers and publicly deployed within Illumina's BaseSpace cloud based computational environment.
Arkas is a two-step cloud pipeline. Arkas-Quantification is the first step, which reduces the computational steps required to quantify and annotate large numbers of samples against large catalogs of transcriptomes. Arkas-Quantification calls Kallisto for on-the-fly transcriptome indexing and quantification recursively for numerous sample directories. Kallisto quantifies transcript abundance from input RNAseq reads by using pseudoalignment, which identifies the read-transcript compatibility matrix4. The compatibility matrix is formed by counting the number of reads with the matching alignment; the equivalence class matrix has a much smaller dimension compared to matrices formed by transcripts and read coverage. Computational speed is gained by performing the Expectation Maximization (EM) algorithm over a smaller matrix.
For RNAseq projects with many sequenced samples, Arkas-Quantification encapsulates expensive transcript quantification preparatory routines, while uniformly preparing Kallisto execution commands within a versionized environment encouraging reproducible protocols. The quantification step automates the index caching, annotation, and quantification associated while running the Kallisto pseudoaligner integrated within the BaseSpace environment. The first step in the pipeline can process raw reads into transcript and pathway collection results within Illumina’s BaseSpace cloud platform, quantifying against default transcriptomes such as ERCC spike-ins, ENSEMBL non-coding RNA, or cDNA build 88 for both Homo sapiens and Mus musculus; further, the first step supports user uploaded FASTA files for customized analyses. Arkas-Quantification is packaged into a Docker container and is publicly available as a cloud application within BaseSpace.
Previous work7 has revealed that filtering transcriptomes to exclude lowly-expressed isoforms can improve statistical power, while more-complete transcriptome assemblies improve sensitivity in detecting differential transcript usage. Based on earlier work by Bourgon et al.8, we included this type of filtering for both gene- and transcript-level analyses within Arkas-Analysis. The analysis pipeline automates annotations of quantification results, resulting in more accurate interpretation of coding and transcript sequences in both basic and clinical studies by just-in-time annotation and visualization.
Arkas-Analysis integrates quality control analysis for experiments that include Ambion spike-in controls, multiple normalization selections for both coding gene and transcript differential expression analysis, and differential gene-set analysis. If ERCC spike-ins, defined by the External RNA Control Consortium9, are detected then Arkas-Analysis will calculate Receiver Operator Characteristic (ROC) plots using 'erccdashboard'10. The ERCC analysis reports average ERCC Spike amount volume, comparison plots of ERCC volume amount, and normalized ERCC counts (Figure 1).
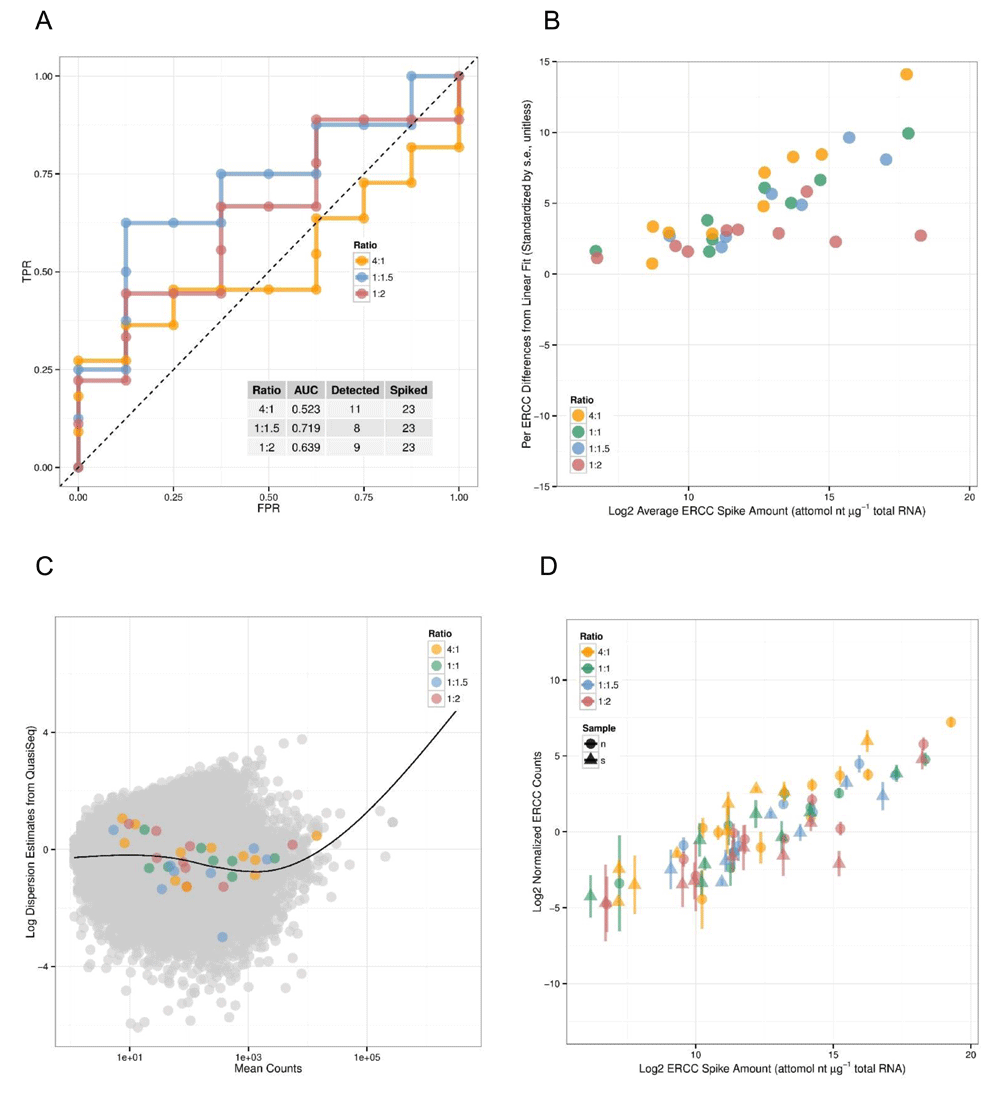
A) The Receiver Operator Characteristic plot. The X-axis shows the False Positive Rate, the Y-axis shows True Positive Rate. B) and D) show the spike-in total RNA amounts with a linear model fit, and quantified ERCC transcript counts. C) shows a dispersion of mean transcript abundance counts and the estimated dispersion.
Subsequent analyses import the data structure from SummarizedExperiment (Morgan, 2016) and create a sub-class titled KallistoExperiment that preserves the S4 structure and is convenient for handling assays, phenotypic and genomic data. KallistoExperiment includes GenomicRanges11, preserving the ability to handle genomic annotations and alignments, supporting efficient methods for analyzing high-throughput sequencing data. The KallistoExperiment sub-class serves as a general-purpose container for storing feature genomic intervals and pseudoalignment quantification results against a reference genome called by Kallisto. By default KallistoExperiment couples assay data such as the estimated counts, effective length, estimated median absolute deviation, and transcript per million count where each assay data is generated by a Kallisto run; the stored feature data is a GenomicRanges object from11, storing transcript length, GC content, and genomic intervals.
Given a KallistoExperiment containing the Kallisto sample abundances, principal component analysis (PCA) is performed12 on trimmed mean of M-value (TMM) normalized counts13 (Figure 2A). Differential expression (DE) is calculated on the library normalized transcript expression values, and the aggregated transcript bundles of corresponding coding genes using limma/voom linear model14 (Figure 3A). Further, an additional PCA and DE analysis of both transcripts and coding genes is performed using in-silico normalization using factor analysis15 (Figure 2B, Figure 3B, Figure 3C). In each DE analysis FDR filtering method is defaulted to 'Benjamini-Hochberg', if there are no resultant DE genes/transcripts the FDR methods is switched to 'none'. Arkas-Analysis consumes the Kallisto data output from Arkas-Quantification, and automates DE analysis using TMM normalization and in-silico normalization on both transcript and coding gene expression in a defaulted two group experimental design, which allows end-users to select the normalization type best suited for their needs.
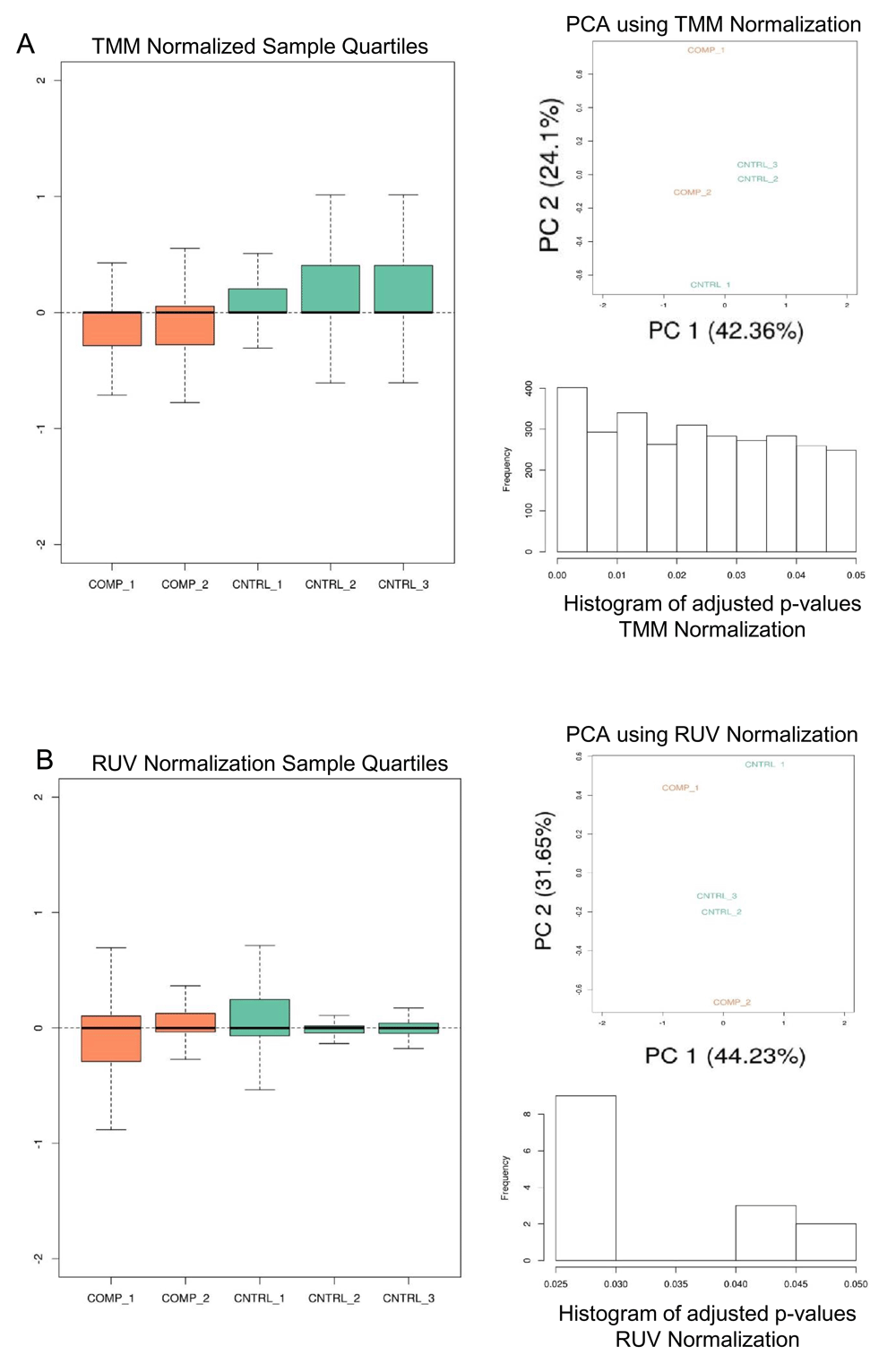
A) TMM normalization is performed on sample data and depicts the sample quantiles on normalized sample expression, PCA plot, and histogram of the adjusted p-values calculated from the DE analysis. Orange is the comparison group and green is the control group. B) A similar analysis is performed with RUV in-silico normalization.
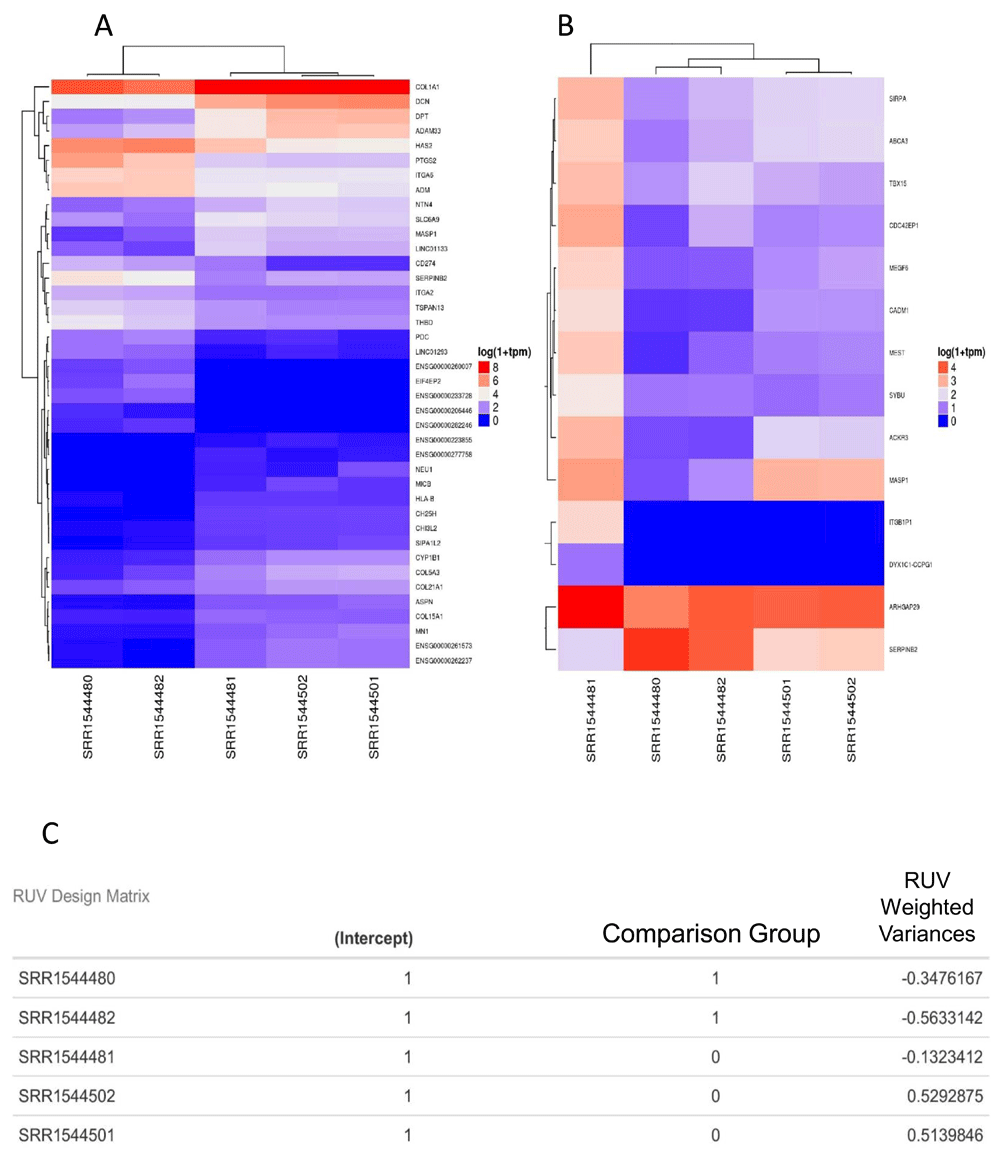
A) DE analysis using TMM normalization. The X-axis is the sample names (test data), the Y-axis are Gene symbols (HUGO). Expression values are plotted in log10 1+TPM. B) Similar analysis using RUV normalization. C) The design matrix with the RUV adjusted weights. The sample names are test data used in demonstrating the general analysis report output.
Gene set differential expression, which includes gene-gene correlation inflation corrections, is calculated using Qusage16. Qusage calculates the variance inflation factor, which corrects the inter-gene correlation that results in high type 1 errors using pooled or non-pooled variances between experimental groups. The gene set enrichment is conducted using Reactome pathways constructed using ENSEMBL transcript/gene identifiers (Figure 4 and Table 1); REACTOME gene sets are not as large as other databases, so Arkas-Analysis outputs DE analysis in formats compatible with more exhaustive databases such as Advaita. The DE files are compatible as a custom upload into Advaita iPathway guide, which offers an extensive Gene Ontology (GO) pathway analysis. Pathway enrichment analysis can be performed from the BaseSpace cloud system downstream from parallel differential expression analysis and can integrate with other pathway analysis software tools.
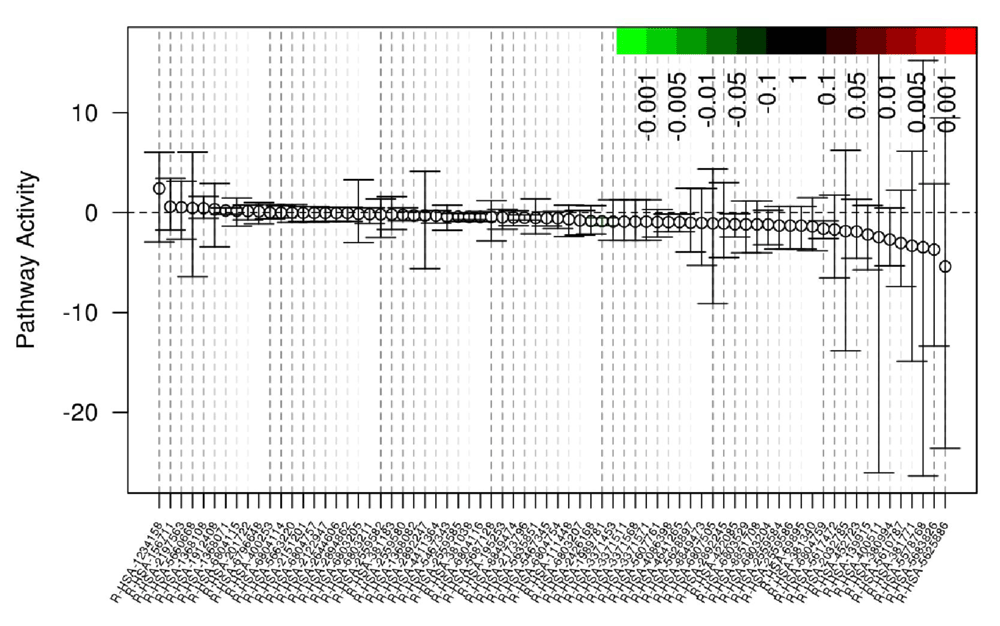
Gene-Set enrichment output report, each point represents the differential mean activity of each gene-set with 95% confidence intervals. The X-axis are individual gene-sets. The Y-axis is the log2 fold change.
The columns represent the Reactome pathway name corresponding to the depicted pathways in Figure 4, the log2 fold change, p-value, adjusted FDR, and an active link to the Reactome website with visual depictions of the gene/transcript pathway. Arkas-Analysis will output a similar report testing transcript-level sets.
| Pathway name | Log fold change | P.value | FDR | Gene URL |
|---|---|---|---|---|
| R-HAS-1989781 | -0.87 | 0.0008 | 0.06 | http://www.reactome.org/PathwayBrowser/#/R-HSA-1989781 |
| R-HAS-2173796 | -0.51 | 0.007 | 0.217 | http://www.reactome.org/PathwayBrowser/#/R-HSA-2173796 |
| R-HAS-6804759 | -1.62 | 0.009 | 0.217 | http://www.reactome.org/PathwayBrowser/#/R-HSA-6804759 |
| R-HAS-381038 | -0.43 | 0.013 | 0.226 | http://www.reactome.org/PathwayBrowser/#/R-HSA-381038 |
| R-HAS-2559585 | -0.4 | 0.032 | 0.341 | http://www.reactome.org/PathwayBrowser/#/R-HSA-2559585 |
| R-HAS-4086398 | -0.95 | 0.033 | 0.341 | http://www.reactome.org/PathwayBrowser/#/R-HSA-4086398 |
| R-HAS-4641265 | -0.95 | 0.033 | 0.341 | http://www.reactome.org/PathwayBrowser/#/R-HSA-4641265 |
| R-HAS-422085 | -1.17 | 0.04 | 0.361 | http://www.reactome.org/PathwayBrowser/#/R-HSA-422085 |
| R-HAS-5467345 | -0.56 | 0.069 | 0.389 | http://www.reactome.org/PathwayBrowser/#/R-HSA-5467345 |
| R-HAS-6804754 | -0.57 | 0.07 | 0.389 | http://www.reactome.org/PathwayBrowser/#/R-HSA-6804754 |
| R-HAS-6803204 | -1.19 | 0.081 | 0.389 | http://www.reactome.org/PathwayBrowser/#/R-HSA-6803204 |
We wished to show the importance of enforcing matching versions of Kallisto when quantifying transcripts because there is deviation of data between versions. Due to updated versions and improvements of Kallisto software, there obviously exists variation of data between algorithm versions (Figure 5, Supplementary Table 1, Supplementary Table 2). We calculated the standardized mean differences, and the variation of the differences between data output from Kallisto versions 0.43 and 0.43.1 (Supplementary Table 2), and found large variation of differences between raw values generated by differing Kallisto versions, signifying the importance of version analysis of Kallisto results.
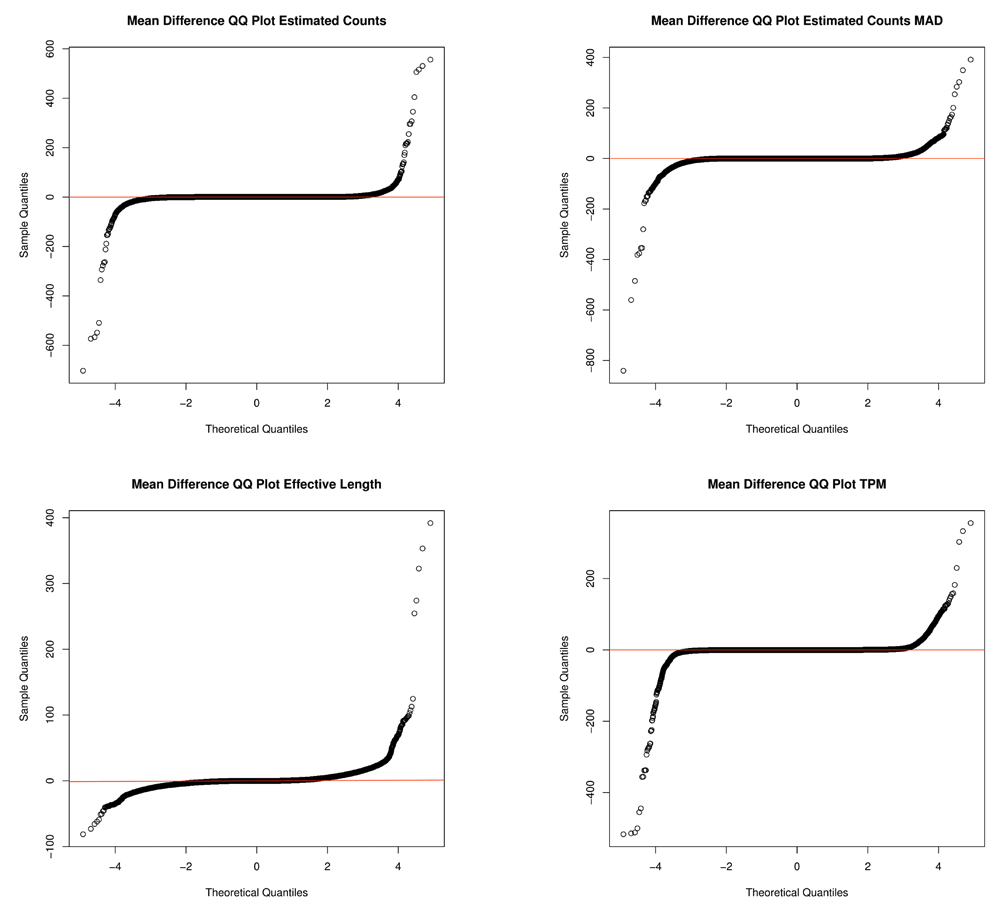
The X-axis depicts the theoretical quantiles of the standardized mean differences. The Y-axis represents the observed quantiles of standardized mean differences.
The Dockerization of Arkas BaseSpace applications versionizes the Kallisto reference index to enforce that the Kallisto software versions are identical, and further documents the Kallisto version used in every cloud analysis. The enforcement of reference versions and Kallisto software versions prevents errors when comparing experiments.
Arkas-Quantification instructions are provided within BaseSpace (details for new users can be found here). The input are RNA sequencing samples, which may include SRA imported reads, and the outputs include the Kallisto data, .tar.gz files of the Kallisto sample data, and a report summary. Users may select for species type (Homo sapiens or Mus musculus), optionally correct for read length bias, and optionally select for the generation of pseudoBAMs. More significantly, users have the option to use the default transcriptome (ENSEMBL build 88) or to upload a custom FASTA of their choosing. For users that wish for local analysis, they can download the sample .tar.gz Kallisto files and analyze the data locally.
The Arkas-Analysis instructions are provided within the BaseSpace environment. The input for the analysis app is the Arkas-Quantification sample data, and the output files are separated into corresponding folders. The analysis also depicts figures for each respective analysis (Figure 1–Figure 4) and the images can be downloaded as a HTML format.
One main advantage of Dockerized analysis software is that it preserves software environments. As an exercise to show the importance of enforcing matching Kallisto versions, we've repeatedly ran Kallisto on the same 5 samples, quantifying transcripts (setting bootstraps=42) against two different Kallisto versions and calculating the standardized mean differences and variation of differences between each run. We ran Kallisto quantification once with Kallisto version 0.43.1, and 4 times with version 0.43.0, merging each run into a KallistoExperiment and storing the runs into a list of Kallisto experiments.
We then analyzed the standardized mean differences for each gene across all samples and calculated the variation of errors for each run quantified using version 0.43.0. Supplementary Table 1 shows the variation of the errors of the raw values such as estimated counts, effective length, and estimated median absolute deviation using the same Kallisto version 0.43.0. As expected, Kallisto data generated by the same Kallisto version had very low variation of errors within the same version 0.43.0 for every transcript across all samples. However, upon comparing Kallisto version 0.43.1 to version 43.0 using the raw data such as estimate abundance counts, effective length, estimated median absolute deviation, and transcript per million values, we found, as expected, large variation of data. Supplementary Table 2 shows that there is large variation of the differences of Kallisto data calculated between versions. Figure 5 depicts the standardized mean differences, i.e. errors, between Kallisto versions fitted to a theoretical normal distribution. The quantile-quantile plots show that the errors are marginally normal, with a consistent line centered near 0 but also large outliers (Figure 5). As expected, containerizing analysis pipelines will enforce versionized software, which benefits reproducible analyses.
The extraction of genomic and functional annotations directly from FASTA contig comments, eliding sometimes-unreliable dependencies on services such as BioMart, are calculated rapidly. The annotations were performed with a run time of 2.336 seconds (Supplementary Table 3) which merged the previous Kallisto data from 5 samples, creating a KallistoExperiment class with feature data containing a GenomicRanges11 object with 213782 ranges and 9 metadata columns. The system runtime for creating a merged KallistoExperiment class for 5 samples was 23.551 seconds (Supplementary Table 4).
The choice of catalog, the type of quantification performed, and the methods used to assess differences can profoundly influence the results of sequencing analysis. ENSEMBL reference genomes are provided to GENCODE as a merged database from Havana's manually curated annotations with ENSEMBL's automatic curated coordinates. AceView, UCSC, RefSeq, and GENCODE have approximately twenty thousand protein coding genes, however AceView and GENCODE have a greater number of protein coding transcripts in their databases. RefSeq and UCSC references have less than 60,000 protein coding transcripts, whereas GENCODE has 140,066 protein coding loci. AceView has 160,000 protein coding transcripts, but this database is not manually curated. GENCODE is annotated with special attention given to long non-coding RNAs (lncRNAs) and pseudogenes, improving annotations and coupling automated labeling with manual curating. The database selected for protein coding transcripts can influence the amount of annotation information returned when querying gene/transcript level databases.
Although previously overlooked, lncRNAs have been shown to share features and alternate splice variants with mRNA, revealing that lncRNAs play a central role in metastasis, cell growth and cell invasion17. LncRNA transcripts have been shown to be functional and are associated with cancer prognosis; proving the importance of studying these transcripts, which are included as defaults within the Arkas pipeline.
Each transcript database is curated at different frequencies with varying amounts RNA entries that influences that mapping rate. GENCODE loci annotations contain 9640 loci, UCSC contain 6056 and RefSeq contain 4888. GENCODE annotations have the greatest number of lncRNA, protein and non-coding transcripts, and highest average transcripts per gene, with 91043 transcripts unique to GENCODE, absent for UCSC and RefSeq databases. ENSEMBL and AceView annotate more genes in comparison to RefSeq and UCSC, and return higher gene and isoform expression labeling improving differential expression analyses18. ENSEMBL achieves conspicuously higher mapping rates than RefSeq, and has been shown to annotate larger portions of specific genes and transcripts that RefSeq leaves unannotated18. Although ENSEMBL has been shown to detect the same differentially expressed genes as AceView, ENSEMBL/GENCODE annotations are manually curated and updated more frequently than AceView18. The choice of transcriptome will definitely influence the power of an analysis, thus Arkas cloud analysis applications use ENSEMBL build 88 (ncRNA, and cDNA) by default for Homo sapiens and Mus musculus and also allow users to upload customized FASTA files.
Reproducible research should consistently link the works developed by the research community to unique data environments such as clinical, sequencing and other experimental data, used in the construction of the published work. The aim for transparent research methodologies is to clearly define their association with every research experiment, minimizing opaqueness between findings and methods. For clinical studies, re-generating an experimental environment has a very low success rate19, which is why non-validated preclinical experiments have spawned the development of best practices for critical experiments. Re-creating a clinical study has many challenges, for example the difficult nature of a disease, the complexity of cell-line models in mouse and human that attempt to capture human tumor environment, and limited power through small enrollments in clinical trials19. Experimental validation is quite difficult and dependent on the skillful performance of an experiment, and an earnest distribution of the analytic methodology, which should contain most, if not all, raw and resultant data sets.
With recent developments for virtualized operating systems, developing best practices for bioinformatic confirmations of experimental methodologies is much more straightforward than duplicating clinical trials' experimental data. Recent technology advancements such as Docker allow for local software environments to be preserved using a virtual operating system. Docker allows users to build layers of read/write access files, creating a portable operating system which exhaustively controls software versions and data, and systematically preserves the complete software environment. Conserving a researcher's developmental environment advances analytical reproducibility if the workflow is publicly distributed. We suggest a global distributive practice for scholarly publications that regularly includes the virtualized operating system containing all raw analytical data, derived results, and computational software. Currently, Docker, compiled software through CMake, and virtual machines are being utilized, showing progress toward a global distributive practice linking written methodologies, and supplementary data, to the utilized computational environment20.
Comparing Docker as a distributive practice to virtual machines seems roughly equivalent. Distributed virtual machines are easy to download, and the environment allows for re-generating resultant calculations. However, this is limited if the research community advances the basic requirements for written methodologies and begins to adopt a large scale virtualized distribution, converging to an archive of method environments which would make hosting complete virtual machines impractical or impossible. If an archive were constructed where each research article would link to a distributed methods environment, then an archive of virtual machines for the entire research community is impossible. However, an archive of Dockerfiles is more realistic because a Dockerfile consists of only a few bytes in size.
Novel bioinformatic software is often distributed as a cross-platform flexible build process independent of compiler, which reaches Apple, Windows and Linux users. The scope of novel analytical code is not to manage nor preserve computational environments, but to have environment independent source code as transportable executables. Docker, however, does manage operating systems, and the scope for research best practices does include gathering sets of source executables into a single collection of minimum space and maximum flexibility. Docker can provide the ability for the research community to simultaneously advance publication requirements and develop the future computational frameworks in cloud.
Another advantage for using Docker as the machine manifesting the practice of reproducible research methods, is that there is a trend of well-branded organizations such as Illumina's BaseSpace platform, Google Genomics, or SevenBridges (all of which offer bioinformatic computational software structures), to use Docker as the principal framework. Cloud computational environments offer many advantages over local high-performance in-house computer clusters, which systematically structure reproducible methodologies and democratize medical science. Cloud computational ecosystems preserve an entire developmental environment using the Docker infrastructure, improving bioinformatic validation. Containerized cloud applications form part of the global distributive effort and are favorable over local in-house computational pipelines because they offer rapid access to numerous public workflows, easy interfacing to archived read databases, and they accelerate the upholding process of raw data. The Google Genomics Cloud has begun to make first steps with integrating cloud infrastructure with the Broad Institute, whereas Illumina's BaseSpace platform has been hosting novel computational applications since its launch.
Scholarly publications that choose only a written method section passively make validation gestures, which is arguably inadequate in comparison to the rising trend or well-branded organizations. We envision a future where published work will share conserved analytic environments, with cloud software accessed by web-distributed methodologies, and/or large databases organizing multitudes of Dockerfiles with accession numbers, strengthening links between raw sequencing data and reproducible analytical results.
Cloud computational software does not only wish to crystallize research methods into a pristine pool of transparent methodologies, but also matches the rate of production of high quality analytical results to the rate of production of public data, which reaches hundreds of petabytes annually. In a talk given by Dr. Atul Butte in December 2015, he discussed that with endless public data, the traditional method for practicing science has inverted; no longer does a scientist formulate a question and then experimentally measure observations that test the hypothesis. In the modern area, empirical observations are being made at an unbounded rate, the challenge now is formulating the proper question (more details on his talk can be found here). Given a near-infinite amount of observations, what is the phenomena that is being revealed? Cloud computational software can accelerate the production of hypotheses by increasing the flexibility and efficiency of scientific exploration.
Many bioinformaticians have noted a rising trend in biotechnology, predicting that open data and open cloud centers will help democratize research efforts and create a more inclusive practice. With the presence of cloud interfacing applications such as Illumina's BaseSpace Command Line Interface, DNA-Nexus, SevenBridges, and Google Genomics becoming more popular, cloud environments pioneer the effort for achieving standardized bioinformatic protocols.
Democratization of big-data efforts has some possible negative consequences. Accessing, networking, and integrating software applications for distributing data as a public effort requires massive amounts of specialized technicians to maintain and develop cloud centers that many research institutions are migrating toward. Currently, it is fairly common for research centers to employ high-performance computer clusters which store laboratory software and data locally; cloud computing clusters are beginning to offer clear advantages compared to local closed computer clusters. Collaborations are becoming more common practice for large research efforts, and sequencing databases have been distributing data globally, making cloud storage more efficient. This implies that services from cloud centers will most likely be offered by very few elite organizations because the large scale of cloud services will prevent incentives for smaller companies.
It is very likely that only a few elite organizations will provide services to cloud computing environments, acting as a gateway which directs the global research community toward a narrow set of well established, standardized, computational applications. With regard to recent changes relating to media consumption and e-commerce, democratization allows independent alternative selections far greater exposure, equalizing profits for lower ranked selections “at the tail", however it may be possible that the abundant amount of data distributed over storage archives, which stimulates an economically abundant environment, could shift into a fiercely controlled economic environment of scarcity. For example, if a gold-standard is reached for computational applications, the range of alternative selections could remain non-existent, which may diminish the future roles of bioinformaticians. This possible scenario suggests bioinformaticians could be re-directed to small garages instead of the technocratic places such as Silicon Valley, motivated not from a spirit of entrepreneurialism, but from a lack of funding.
Automative downstream analyses is not without its drawbacks; most computational software is highly specialized for niche groups with a mathematical framework constructed by specialized assumptions, this may require a diverse array of computational developments, and thus a large community of developers. The automation of analytical results seems almost unavoidable, and the benefits seem to outweigh the negative consequences.
Arkas integrates the Kallisto pseudoalignment algorithm into the BaseSpace cloud computation ecosystem that can implement large-scale parallel ultra-fast transcript abundance quantification. We reduce a computational bottleneck by freeing inefficiencies from utilizing rapid transcript abundance calculations and connecting accelerated quantification software to the Sequencing Read Archive. We remove the second bottleneck because we reduce the necessity of database downloading; instead we encourage users to download aggregated analysis results. We also expand the range of common sequencing protocols to include an improved gene-set enrichment algorithm, Qusage, and allow for exporting into an exhaustive pathway analysis platform, Advaita, over the AWS EC2 field in parallel.
Controls: SRR1544480 Immortal-1
SRR1544481 Immortal-2
SRR1544482 Immortal-3
Comparison: SRR1544501 Qui-1
SRR1544502 Qui-2
Latest source code:
https://github.com/RamsinghLab/Arkas-RNASeq
Archived source code as at the time of publication:
License:
MIT license
For Homo-sapiens and Mus-musculus ENSEMBL FASTA files were downloaded here for release 88.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)