Keywords
Variant calling, Whole genome sequencing, Structural variation,
Variant calling, Whole genome sequencing, Structural variation,
Genomic structural variants (SVs) are defined as large genomic rearrangements and consist of inversion and translocation events, as well as deletions and duplications1. SVs have been shown to be both the direct cause and a contributing factor in many different human genetic disorders, as well as in more common phenotypic traits2–4.
In genetic diagnostics, current techniques such as FISH5 and microarray studies6 have limited resolution, and the information obtained often needs to be complemented by additional methods for a correct interpretation5,6. Previous studies have shown that whole genome sequencing can be used to successfully identify and characterize structural variants in a single experiment7.
The advent of systems like the Illumina HiSeqX allows researchers to sequence Whole Human Genomes (WHG) at high (i.e. 30X) coverage and at a relatively low cost (i.e. 1000$ per WHG)8. The ability to produce large amounts of data at an unprecedented speed has initiated a flourishing of computational tools that are able to identify (i.e. call) structural variants and/or chromosomal breakpoints (i.e. the exact position in the chromosome at which SV takes place). These tools are commonly called variant callers, or simply, callers. Variant callers generally require an alignment file (in BAM format) as input and try to identify differences between the reference genome and the donor/patient genome. To detect structural variants, callers generally use heuristics based on different signals in the WGS data. These signals include discordant pairs9,10, read-depth11, and split-reads12. Some callers try to reconstruct the patient sequence by applying either local13 or global14 de novo assembly techniques. Depending on the size, variant type, and characteristics of the sequencing data, the most suitable method for detecting a variant will differ15.
Thanks to the ability to produce high quality sequencing data at a relatively low cost, as well as the potential to detect any variant from a single experiment, whole exome sequencing16,17 and whole genome sequencing7,18 could be highly useful in clinical diagnostics, especially to study rare disease causing variants. However, to avoid high validation costs, highly precise, yet sensitive callers are needed. To further complicate the situation, an abundance of sequencing platforms and library preparation methods are available19. Sequencing data generated from these different sources have different properties, such as read length and coverage20. As an example, it has been shown that large insert mate pair libraries are well suited to detect SVs21, mainly due to the ability to span repetitive regions and complex regions that act as drivers of structural variation22 and due to the sensitivity derived from a large physical span coverage compared to small insert size sequencing coverage.
Here we present a new variant caller, TIDDIT. The name highlights the ability to detect many different types of SVs; including but not restricted to translocations, inversions, deletions, interspersed duplications, insertions and tandem duplications. TIDDIT utilizes discordant pairs and split reads to detect the genomic location of structural variants, as well as the read depth information for classification and quality assessment of the variants. By integrating these WGS signals, TIDDIT is able to detect both balanced and unbalanced variants. Finally, TIDDIT supports multiple paired-end sequencing library types, characterized by different insert-sizes and pair orientations.
To simplify the search for rare disease causing variants, TIDDIT is distributed with a database functionality dubbed SVDB (Structural Variant DataBase). SVDB is used to create structural variant frequency databases. These databases are queried for rare disease causing variants, as well as variants following certain inheritance patterns. Utilizing the database functionality, the analysis of rare variants may be prioritized, thus speeding up the diagnosis of rare disease causing variants. To our knowledge, no available caller provides such an extensive framework to call and evaluate rare disease causing structural variants.
Detection of structural variants. TIDDIT requires a coordinate-sorted BAM file as input. There are two phases; in the first phase, coverage and insert size distribution are computed from the BAM file. These data will be used in the subsequent phase. In the second phase, TIDDIT scans the BAM file for discordant pairs and split reads and uses these signals to detect and classify structural variants. These two signals are pooled together by redefining each signal as two genomic coordinates Si = (, ), for a split read the position is given to the position of the primary alignment, and the position is given to the position of the supplementary alignment of that read. On the other hand, for discordant pairs, the position is given to the the read having the smallest genomic coordinate, and the position is given to the position of the read that has the largest genomic coordinate. A read pair is deemed to be discordant if the reads map to different contigs, or if the distance exceeds a threshold distance, Td. By default, Td is set to three times the standard deviation plus the average value of the insert size distribution. Every time a SV signal is identified, TIDDIT switches from reading-mode to discovery-mode. As soon as the signal Si = (, ) is identified, the set D is initialized:
At this point TIDDIT searches for other evidence in the neighborhood. Every time a new signal is identified it is added to set D. The construction of this set is halted only when no other signal is identified in W consecutive positions. In more detail, if () = cj (i.e., is a position j on chromosome c), then D will contain the following signals:
The first condition guarantees that D will contain only signals found after position cj, i.e. the position of the first signal within the set D. The existential clause guarantees that the set D will not contain any signal that is too far away from the signals within the set D. D is obtained by reading the ordered BAM file and populating a data structure with all the detected discordant pairs and split reads. If no signal is identified after reading W positions from the last position containing a read added to D, the discovery phase is closed. TIDDIT also records information about local coverage and read orientation while constructing D. Once D is computed, TIDDIT partitions it into distinct sets D1, D2, …, Dk such that:
In other words, D is divided into non overlapping partitions, Dk requiring that all p2 positions form a cluster with properties analogous to the p1 positions. Once D is divided into partitions, TIDDIT checks if any partition represents a structural variant or if it is only noise. In the case of it being a structural variant, TIDDIT tries to associate the identified signal with a specific type of variation. A set Dk is discarded (i.e. the SV is not reported) if the number of pairs forming the set (i.e. the cardinality of the set) is below a given threshold. This threshold is used to control the sensitivity and specificity of TIDDIT. In general, the number of discordant pairs is dependent on multiple factors, and may vary considerably throughout the genome. Therefore the user may need to fine tune the required number of discordant pairs based on the downstream analysis. All callable structural variants in D are reported, and thereafter, D is discarded. TIDDIT will then return to read mode, starting with the next available read pair.
Classification of structural variants. TIDDIT identifies candidate variations using discordant pairs and split reads. To determinate the variant type, TIDDIT analyses read orientation, as well as the coverage across the region of the first reads, second reads, and the region in between. TIDDIT characterizes three levels of coverage: low, normal and high. If is the average coverage computed over the entire genome sequence, then the coverage across a region, C is deemed normal if it is satisfying the following condition:
P ← The ploidy of the organism
If the coverage across a region is lower than normal, it is classified as low coverage. Likewise, if the coverage is higher than normal coverage, it is classified as a high coverage region. The patterns of the variants detected by TIDDIT are represented in Figure 1.
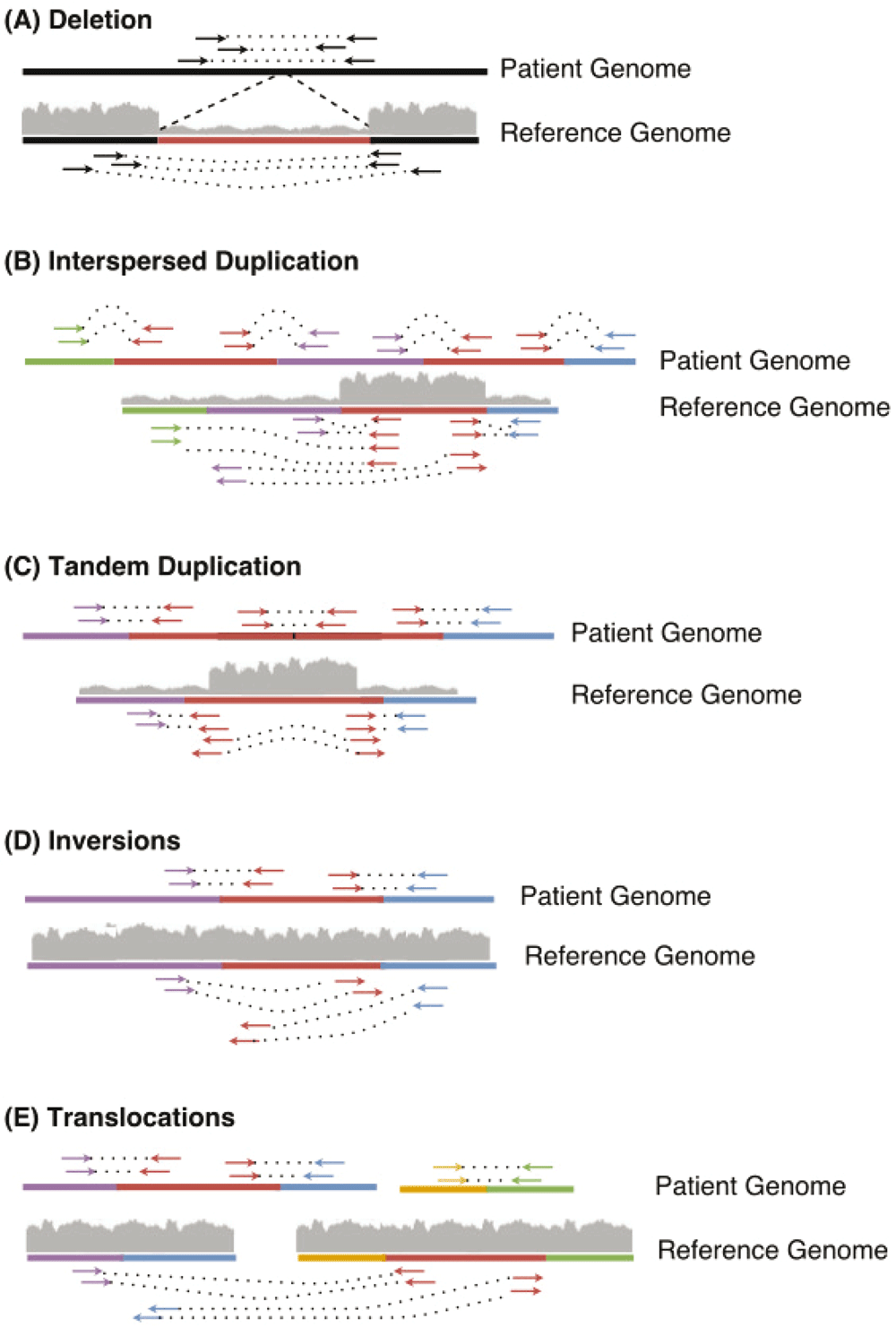
Thereafter, the read orientation and coverage is used for classification of the SV. Deletions (A) are characterized by low coverage across the deleted region (shown in red), and normal orientation. Interspersed duplications (B) are characterized by high coverage across the duplicated region (shown in red), but normal coverage across the other regions affected by the SV. Tandem duplications (C) are characterized by high coverage across the duplicated region (shown in red), the entire duplicated region is found between the discordant pairs/split reads (shown in red), the read pair orientation is inverted compared to the expected library orientation. Inversions (D) are characterized by the orientation of the read pairs (red-purple and red-blue) bridging the inverted region (shown in red). When aligning these discordant pairs to the reference, the reads of the discordant pairs will have the same orientation, forward/forward or reverse/reverse. For split reads, the orientation of the secondary alignment will differ from the primary alignment. Lastly, translocations (E) are characterized by the read pairs bridging the translocated region (red) and its surroundings (blue and purple). Either these regions belong to another contig than the translocated region, or to the same contig but, in contrast to interspersed duplications, the coverage across the translocated region is normal.
In a deletion event (Figure 1A) a region is absent in the patient genome but present in the reference. When aligned to the reference, the read pairs flanking the deleted region will have a larger insert size than what is expected based on the library insert size distribution. Moreover, split reads will be formed in such a way that one part of the read is aligned to one side of the breakpoint, and another part of the read will be aligned to the other side of the deletion. Furthermore, the coverage of the lost region will be lower than expected. Therefore, TIDDIT will classify a variant as a deletion if the region flanked by the discordant pairs/split reads is classified as a low coverage region.
In an interspersed copy number event (Figure 1B), an extra copy of a region within the genome (the red sequence in Figure 1B) is positioned distant from the original copy. In this case, there will be read pairs and reads bridging the duplicate copy and the distant region. When aligned to the reference, the read pairs will appear to have unexpectedly large insert size, and the reads will appear split across the reference. Thus, TIDDIT will detect these signals. Furthermore, the coverage of the copied region will be higher than expected. By scanning the coverage of the regions where the reads of the discordant pairs are located, TIDDIT will find which region has an extra copy. The interspersed duplication is reported as two events, an intrachromosomal translocation between the duplicated region and the position of the copy, and a duplication across the duplicated region.
In a tandem duplication event (Figure 1C), the extra copy is positioned adjacent to the original copy. Since the distance between the segments is small, there will be pairs where one read is located at the end of the original copy, and the other read is located at the front of the duplicate copy (Figure 1C). When aligned to the reference, the insert size of these read pairs will be as large as the tandem copy itself (Figure 1C). Similarly, there will be reads bridging the two copies. These reads will appear split when mapped to the reference. Furthermore, the orientation of these read pairs and split reads will be inverted. Moreover, the coverage across the entire genomic region will be higher than expected. Thus to classify tandem duplications, TIDDIT will search for sets of discordant pairs/split reads that have inverted read pair orientation as well as high coverage between the read pairs.
In an inversion event (Figure 1D), the sequence of a genomic region is inverted. When aligning the sequencing data the insert size of the read pairs bridging the breakpoints will appear to be larger than expected. Furthermore, both read pairs will get the same orientation, such as reverse/reverse or forward/forward. TIDDIT employs the read pair orientation in order to identify inversions. Given that the inversion is large enough, TIDDIT will find the pairs bridging the breakpoints of the inversion, and will classify the variant as an inversion if the orientation of the discordant pairs are forward/forward or reverse/reverse orientation, and if the orientation of the primary/secondary alignments are forward/reverse, reverse/forward.
In an interchromosomal translocation event (Figure 1E), the first read and second read will map to different contigs (Figure 1E). Reads bridging the translocated segment will appear split between these two contigs. Any read pair mapping to two different contigs is counted as a discordant pair, and any set of signals mapping to different contigs will be classified as an interchromosomal translocation. Intrachromosomal translocation events are similar. They are balanced events, where a genomic region has been translocated to another location within the same chromosome. When aligned to the reference region, these variants will give rise to signals where one read is mapping to the translocated region, and the other read mapping to the region where the translocated region is positioned. This will give rise to pairs having larger insert size than expected. However, since there is no change in copy number, the coverage will be normal across the discordant pairs. The orientation of the reads forming the discordant pairs will depend on whether the translocated region is inserted in its original orientation, or if it is inverted relative to its original orientation. Thus the read pairs may retain the standard library orientation, but the orientation could also be inverted. Therefore, intrachromosomal translocations are classified by scanning for discordant pairs having either forward/reverse or reverse/forward orientation and normal coverage in the region between those reads.
Filtering of structural variant calls. For each called variation, several statistical values are calculated. They serve two purposes: to provide more information and to filter out noise. In the former case, statistics are employed to understand the structure of the variant and to relate it to the rest of the genome. In the latter case, filters are employed to improve the precision of TIDDIT. TIDDIT utilizes four complementary filters: Expected_links, Few_links, Unexpected_coverage, and Smear. These heuristics are used to set the FILTER column of the VCF generated by TIDDIT.
The main goal of Expected_links is to filter variants caused by random events such as contamination or sequencing errors. It uses a statistical model to compute the expected number of discordant pairs23 using the library insert size, read length, ploidy of the organism and coverage across the region affected by the structural variant. A variant that is defined by less than 40% of the expected number of pairs will fail the Expected_links quality test, and is set to Expected_links in the FILTER column of the VCF. The statistical model supports variants that are called using discordant pairs, hence for calls based on split reads exclusively, the number of split reads divided by coverage and ploidy is used as an estimate of the expected number of split reads.
Few_links aims to filter out calls that are caused by reference errors and misalignment of repetitive regions. As mentioned previously, a variant is defined as a set of positions, In order to compute the Few_links filter, for each Dk, TIDDIT creates another set called DSpurr, containing spurious read pairs. Spurious reads pairs are pairs that belong to the interval identified by Dk, but whose mates align to a different chromosome from the one where the pairs form Dk. In other words, TIDDIT checks if a genomic location where a SV can be called is linked to multiple other events. In this case the suspicion is that the called SV is the consequence of a repetitive element. If the fraction of spurious read pairs is too high, the variant within Dk is considered unreliable, and thus its filter flag is set to Few_links. The fraction of spurious read pairs is considered too high if the following formula holds true:
P ← The ploidy of the organism
Smear is a filter designed to remove variants called due to large repetitive regions. In these regions, the split reads and discordant pairs will map in a chaotic manner, hence these regions may appear to be affected by large structural variation. These calls are recognized by searching for variants were the regions of the first read and its mate overlap, or where the regions of the primary and secondary alignment overlap. If this is the case, the variants will fail the Smear test. The filter flag of variants that fail this test is set to Smear.
Lastly, Unexpected_coverage also aims to filter out calls caused by reference errors and misalignments, but unlike the Few_links, it employs coverage information. The Unexpected_coverage filter uses the coverage across the region of the p1 signals as well as the region of p2 to determine the quality of a variant call. If the coverage of any of these regions is 10 or more times higher than the average library coverage, the variant will fail the Unexpected_coverage test, and its FILTER column is set to Unexpected_coverage.
Any variant that passes these filters is set to PASS, those that fail are set according to the filter that rejected the variant. By removing all variants that did not pass this quality control, the precision of TIDDIT improves considerably.
Using the structural variant frequency database (SVDB) software, the user may compare variants found in different samples and annotate the VCF files with the frequency of each variant. The frequency database is built with multiple VCF files containing structural variant information. The VCF files may be generated using any caller that reports structural variants according to the VCF standard. By removing high frequency variants from the VCF file, rare variants may be detected. The database could also perform trio analysis and filter out variants following a certain frequency pattern within a family.
The database is an SQLite database. It contains one entry per input variant. These entries describe the genomic position, variant type, and sample id of the variant. Moreover, each variant is given a unique index number. These SQLite databases may then be used either directly to provide frequency annotation, or they may be exported to a VCF file.
Export. The variants of the SQLite structural variant database may be exported to a VCF file. The VCF file is a standard structural variant VCF file, generated by clustering similar variants within the SQLite database, and reporting each cluster as a single VCF entry. The INFO field of each VCF entry contains four custom tags. The FRQ tag describes the frequency of the variant, the OCC tag describes the total number of samples carrying the variant, the NSAMPLES tag describes the total number of samples within the database, and lastly, the variant tag describes the position and id of each clustered variant.
The clustering of the variants is performed using one out of two methods, either an overlap based clustering method or DBSCAN24.
Annotation. The main purpose of the structural variant frequency database is to query it and use it for frequency annotation. The frequency database is queried using a VCF file. All variants within the VCF file will be annotated with the frequency of that variant within the frequency database. The database used for querying may either be an SQLite database, an exported VCF file, or a multi-sample VCF file such as the thousand genome structural variant VCF.
If an SQLite database is chosen, two separate algorithms are available, overlap or DBSCAN. If DBSCAN is chosen, all variants of the SQLITE database and the query VCF are clustered using DBSCAN. Thereafter, the frequency of each query variant is set to the number of separate samples represented in the cluster of that query variant.
Overlap based clustering. The most critical part when building and querying the database is to determine if two SVs represent the same event or not. When using the overlap based clustering algorithm, two interchromosomal variants are considered equal if the distance between their breakpoints does not exceed a certain distance threshold. This distance is set to 5 kilobases (kB) as default. However, the user may change it to suit any kind of data. Secondly, for intrachromosomal variants fulfilling the breakpoint distance criteria, the overlap O is computed and used to determine if the variants are similar enough. For a given chromosome, to compute O each variant var is regarded as an ordered set of genomics coordinates:
var = {i, i + 1,..., j — 1, j}
The overlap parameter O is defined as the cardinality of the intersection of two variants, divided by the cardinality of the union of the same overlapping variants:
Where var1 and var2 are two overlapping variants (O equals to 0 if the variants are not overlapping). The default threshold value of the overlap parameter is 0.6.
The database software extracts the variant type from the ALT field. By default, variants of different type will not be considered equal even if the overlap is high enough. When constructing the database, the variants of the VCF file are transferred into an SQLite database. This database can then be queried directly using the previously described procedure. The database could also be exported into a VCF file.
When exporting the database, variants from different patients overlap in complex patterns and form chains where not all variants in one chain are overlapping according to the user set parameters. Consider three variants, varA, varB, and varC as an example;
The overlap between variants varA and varB aswell as varA and varC satisfies the overlap settings, O. However, the overlap between varC and varB is not big enough:
To resolve these cases, all the variants, V found in all patients are divided into sets χ based on the overlap threshold, O such that
Each Vi in χi may not have a high enough overlap with all Vj in χi , instead the variants in χi may form complex chain-like patterns of overlap. To resolve these chains of variants, each χi is divided further into subsets of variants, , such that
Each set is represented by one single variant, . And for each variant Vk in , the overlap threshold of is satisfied.
The sub clusters are computed by defining two sets, A and B.
A = B = χi
is picked at random from A, and added to . Thereafter, is tested for overlap against each variant within B. A variant Vk in B will be added to if it satisfies the overlap threshold. Once the overlap is calculated between and each Vk in B, A is redefined as A \ = A, thereafter is printed as a database entry, and the frequency of is set to the number of patients carrying a variant found in . If the set A is non-empty, a new is initiated and another is sampled from A, and the described process is repeated again.
DBSCAN clustering. The second clustering algorithm used by SVDB is DBSCAN. DBSCAN requires two parameters, epsilon and minPTS24. At default epsilon is set to 500 bases, and minPTS is set to 2 samples. However, these parameters may be changed by the user.
The DBSCAN clustering is performed by dividing each chromosome and variant type into separate sub-databases; thereafter, a 2-dimensional coordinate system is defined for each sub-database. For intrachromosomal variants, the x coordinate of this plane corresponds to the start position of the variant, and the y coordinate within the plane corresponds to the end position of the variant.
Interchromosomal translocations involve two chromosomes, so when clustering these variants, the contig id is sorted according to lexicographic order. Out of the two contigs involved in the rearrangement, the contig ordered first is set to the x axis, and the contig last in order is set to be the y axis. Thereafter, each variant is added to the plane as described for intrachromosomal variants. This procedure is repeated for any possible chromosome pair, and each variant type on each chromosome pair.
Once each plane is defined, the variants within each separate plane are clustered using DBSCAN.
Detection of structural variants. TIDDIT requires a coordinate sorted BAM file as input, and may be run in two separate modes; variant calling or coverage computation. The coverage computation mode is used to compute the coverage across the entire genome, and returns a BED file as output. The variant calling mode is run to analyse SV across the entire genome. All the detected SVs are returned in a single VCF file.
System requirements. TIDDIT has been tested on a large number of datasets; in general, TIDDIT will perform variant calling in less than 2 hours(3) using a single CPU core, and 2 gigabytes of RAM memory or less. The time consumption and memory usage is mainly dependent on the coverage and quality of the input data.
TIDDIT has been tested on Linux as well as Apple macOS. TIDDIT is easy to install and requires only standard c++ libraries. TIDDIT is installed using cmake (https://cmake.org/) and make (https://www.gnu.org/software/make/).
Downstream analysis. TIDDIT generates a VCF file containing structural variant calls across the entire genome. Initially, these calls may be filtered based on the quality filters described in the implementation section, as well as the SVDB software, using either internal samples or an external dataset such as thousand genomes structural variants25 (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/integrated_sv_map/). The TIDDIT output VCF is compatible with any tool which supports VCF, hence there’s a large number of tools available for further analysis of the variant calls. Typically, the VCF file is annotated using software such as VEP26 or snpEFF27. Tools such as VCFtools28 and BEDtools29 may be used to remove low quality calls, or filter calls within specific genomic regions.
The performance of TIDDIT was evaluated using simulated, as well as public datasets containing a large number of validated variants of different size and type. The performance of TIDDIT was compared to current top of the line callers, including Lumpy30, Delly10, CNVnator11, Manta9, and Fermikit14. These are well known callers that have been tested throughout numerous projects.
Three separate datasets were used to evaluate TIDDIT. First, a simulated dataset generated using Simseq31 and SVsim (https://github.com/gregoryfaust/svsim) was used. Next, two public large scale sequencing datasets were used, namely the NA12878 sample, and the HG002 sample32. Truth sets of validated variants were found for each of the public datasets32,33. A detailed description of the benchmarking of TIDDIT is given in Supplementary File 1.
To test the performance of the callers, the tools were run on the simulated dataset. The dataset consists of four separate samples, one per variant type. The coverage of these simulated samples was set to 25X, and the read length and insert size was set to 100 and 350 bp, respectively. For each variant type (deletions, duplications, inversions, translocations), 6000 variants were simulated. Hence, the simulated dataset contains 24000 known variants of different type. The results are presented in Table 1. For each caller the sensitivity and precision was computed. The sensitivity differs between different callers and variant types. Table 1 shows that Fermikit is the caller with the lowest sensitivity. TIDDIT, however, is consistently the caller with the highest sensitivity. It is important to notice how the high sensitivity of TIDDIT is coupled with extremely high precision. In other words, TIDDIT is not only able to call all the real variation in the dataset, but it is also one of the tools least affected by false positives. For instance, looking more closely at the translocations in Table 1 we can see that TIDDIT has the best trade-off between sensitivity and precision, revealing the highest sensitivity out of all callers, and precision that was only 0.02 lower than Delly and Manta.
The variants were simulated using SVsim and Simseq.
Lastly, to complement the benchmarking on the simulated dataset, and to test TIDDIT on a more diverse set of sequencing data, the same callers were run on the NA12878 and HG002 samples32. The results are presented in Table 2. We computed both sensitivity and precision, in three size intervals. Events 0–100 bp in size are usually not considered structural variation, but they were kept as a size interval anyway since some callers also detect these variants.
These public datasets contain validated deletions of various sizes.
With the NA12878 sample, the performance differed widely between callers and variant sizes (Table 2). With variants larger than 1000 bp, TIDDIT had the highest detection rate and second best precision. FermiKit was the most precise tool, but it must be noted that such result was achieved at the expense of sensitivity, since Fermikit had almost half the sensitivity of TIDDIT. In other words, FermiKit was not able to call many true variants, but the variants that were called were more likely to be correct. On the other hand, TIDDIT was able to call almost all the validated large ( i.e. ≥ 1000 bp) variants, but many calls done by TIDDIT did not overlap with the validated ones. However, since the truthset only contains high quality deletion calls, these non-overlapping calls are not necessarily incorrect. With medium (i.e., 100−1000 bp) and small (i.e., ≤ 100 bp) size variants the performance of the callers differed greatly. For these variants, Manta had the highest sensitivity, while TIDDIT had the highest precision.
Moving on to the HG002 sample, it was found that most variant callers performed worse than with the NA12878 sample. No variant caller produced any significant number of true positive calls in the range of 0–100 bp. TIDDIT had the highest sensitivity on variants larger than 100 bases, and was one of the most precise callers (Table 2).
The computational performance of the six tools was also determined. Despite not being the most important parameter to consider, CPU time rising in importance, especially if one needs to run analysis in a Compute Infrastructure as a Service (e.g. Google Cloud or Amazon). The CPU hour consumption of the callers was measured while analyzing each sample. The results of the measurements are presented in Table 3. During the analysis, each caller except Fermikit was run on a single core of an Intel Xeon E5-2660 CPU, and Fermikit was run using 16 Intel Xeon E5-2660 CPU cores.
It was found that CNVnator and TIDDIT are the most efficient callers, while FermiKit is by far the most expensive caller to run.
Each caller except Fermikit was run on a single core of a Intel Xeon E5-2660 CPU. Fermikit was run on 16 CPU cores. The CPU hour consumption of the Simseq data is reported as the median time consumption across the four Simseq samples.
| CPU hour consumption on SV calling | |||
|---|---|---|---|
| Caller | NA12878 | HG002 | Simseq |
| TIDDIT | 2 | 1 | 1 |
| CNVnator | 2 | 1 | 1 |
| Delly | 30 | 15 | 7 |
| FermiKit | 640 | 120 | 15 |
| Lumpy | 45 | 2 | 7 |
| Manta | 3 | NA | 1 |
Evaluation of the database functionality. The performance of the database functionality was evaluated by building databases of different sizes. These databases were built by randomly sampling individuals from a set of 209 that were sequenced through the thousand genomes project25. These individuals are listed in Supplementary File 1.
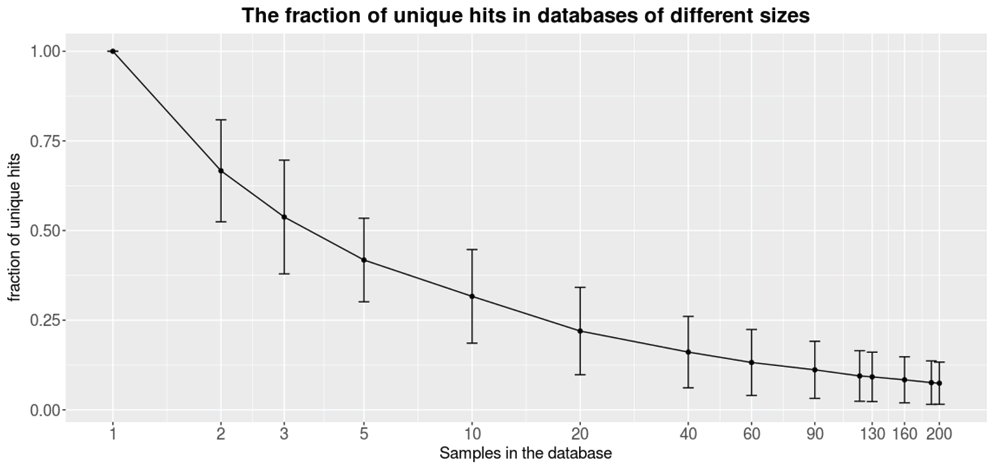
Figure 2 shows how the fraction of unique hits within the database gets lower as the size of the database increases, therefore improving the ability to find unique variants for new patients. A unique hit is defined as a variant that has only been found in the query itself. Already, a relatively small database filtered out a significant amount of variants. On average, a sample queried against a database consisting of only 10 samples contain about 25% unique variants. Still, a larger database filters out more variants. Each query sample was found to contain 7.5% (i.e., ∼250) unique structural variants when filtered against a database containing 200 samples. Since each caller reports a relatively large fraction of false positives (Table 2), the frequency database is necessary to reduce the number of variants. Moreover the frequency database can be use to filter out recurring technical errors connected to the library preparation, sequencing chemistry and alignment of the sequencing data.
The structural variant databases may also be used to benchmark different tools and settings, as well as to compare the SVs within a family or population. As an example, the database functionality was used to study differences between populations sequenced through the thousand genomes project25. Three different populations were selected; Han Chinese from Bejing (CHB), Japanese from Tokyo (JPT), and Yoruba from Ibadan (YRI). In total, 25 samples were analysed; 10,5, and 10 samples from the CHB, JPT, and YRI populations, respectively. All of these samples were analysed using TIDDIT. The similarity of the samples was determined by creating one database per sample, and querying each sample against each database. The fraction of similar SVs was determined by computing the number of common/similar SVs divided by the total number of SVs in that sample. A more detailed description of the analysis and the selected samples is given in Supplementary File 1. It was found that the three populations are distinct based on their SVs. Furthermore, the CHB and JPT populations are relatively similar compared to the YRI population (Figure 3). The CHB population appears to be more homogeneous than the other two populations, with the exception of one individual which appears to be similar to the JPT individuals. On the other hand, the YRI population appears to be most diverse population, and is divided into two clear subpopulations.

These individuals were sequenced through the thousand genomes project. The heatmap is coloured based on the similarity between individuals, and shows that the populations can be distinguished based on their SVs.
Six structural variant callers were benchmarked on simulated data generated by simseq (Table 1), and two public datasets, including NA12878 and HG002 (Table 2). Compared to the other callers, TIDDIT performs well on SV larger than 1000 bp (Table 1, Table 2). TIDDIT is able to identify large structural variants in many experimental setups: low or high coverage, short or long fragment size (i.e., Paired End or Mate Pair) (Table 1, Table 2). Furthermore, TIDDIT has a good balance between sensitivity and precision. Despite being one of the most sensitive tools, it is also one of the most precise tools. These two characteristics (high sensitivity and high precision) in conjunction with a low computational demand makes TIDDIT one of the most capable tools available today for the identification of large SVs greater than 1 kbp from WGS data. TIDDIT does not perform well on small variants (Table 2), however TIDDIT performs really well on large variants, especially balanced variants (Table 1). Since TIDDIT is efficient, produces high quality variant calls, and performs well in multiple settings, TIDDIT could be a great addition to WGS pipelines.
Even though the presented benchmarking is extensive, it is not fully complete. The public datasets lack large enough truth sets for balanced variants and duplications. Since the callers perform differently on different variants (Table 1), it would be of value to benchmark the callers against a more varied set of variants. Moreover, the variants of these truth sets are generally small. For instance, the median size of the deletions of NA12878 is about 250 bases, which is smaller than the traditional size of structural variation3.
The Human genome contains a large amount of repetitive regions, and each individual carries a large number of structural variants34,35. Due to these reasons, the number of detected structural variants per samples is generally high. Thus, finding a rare disease-causing variant among such a large number of common variants is difficult and time consuming. TIDDIT is distributed together with structural variant database software. This software package uses structural variant VCF files to construct variant frequency databases. The annotation provided by these databases is then used to filter common variants as well as reference errors.
By filtering out high frequency variants from the VCF file, rare disease causing variants can easily be detected (Figure 2). Moreover, the database could be constructed to follow variants that all samples have in common, such as an inherited disease variant within a family, or known disease causing variants, as well as to search for variants following a certain inheritance pattern, or to compare the SVs of different populations (Figure 3). The structural variant databases could also be used to benchmark different software tools, settings, and library preparation methods. The database functionality can be used for results obtained from any tool that generates a valid VCF file as format.
TIDDIT is an efficient and comprehensive structural variant caller, supporting a wide range of popular sequencing libraries. Not only does TIDDIT have the functionality of a structural variant caller, it also has a set of functions that helps the user perform further analysis of the bam file. These functions include depth of sequencing coverage analysis and structural variant database functionality. By utilizing these functions, TIDDIT could either perform advanced analysis on it’s own or be used to perform a wide range of tasks within a variant analysis pipeline. TIDDIT has already been employed in many studies and demonstrated its potential not only with the commonly used Nextera mate pair libraries from Illumina7,18,36 but also with the TrueSeq Nano and PCR-free Paired End libraries37.
Latest source code: https://github.com/J35P312/TIDDIT
Archived source code as at the time of publication: http://doi.org/10.5281/zenodo.43951738
License: GNU General Public License version 3.0 (GPLv3)
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)