Keywords
Potassium ion, sodium ion, ion pairing, molecular dynamics, ion coordination
Potassium ion, sodium ion, ion pairing, molecular dynamics, ion coordination
Salt solutions are the natural environment in which biological molecules act. Moreover, the dissolved ions themselves largely participate in many biological processes on a molecular level. Metal ions are essentially cofactors of many enzymes and may coordinate with charged groups, thus modulating protein-protein interactions and their activity1. Many of these manifestations are due to specific ion coordination with charged groups on protein surfaces and other counterions in solutions, rather than the alteration of the aqueous solution structure in the bulk2. Such ion - counterion pairing has been validated experimentally, and described theoretically using molecular simulations3.
Despite apparent similarity, the biological roles of sodium and potassium are very different. For example, the potassium to sodium ion concentration ratio is high inside the cell and low outside, which gives rise to membrane potentials. These two vital ions also demonstrate different catalytic capacity in the model reaction of prebiotic peptide synthesis, where potassium shows a higher activity4. In addition, their roles in abiogenesis are of high interest5.
It was supposed that sodium binds to charged groups on protein surfaces more strongly than potassium does, which probably correlates with the "salting out" sodium effect on proteins, and "salting in", as is known for potassium6. Using an X-ray absorption study of solutions containing dissolved ions and acetate or glycine molecules, it was demonstrated that sodium has superior affinity to carboxylate, one of the major anionic groups in proteins7. In a number of works, such a difference was explained using a combination of molecular dynamics and ab initio calculations6–9. With this method, the difference between sodium and potassium ion association free energies with carboxylate groups has been calculated9.
Using molecular dynamics, it was demonstrated that not only direct ion - carboxylate pair but also the solvent shared ion - carboxylate paired configurations are of great importance10–12. Particularly, these solvent-mediated states appear more populated than direct contact ion pair, and can determine the thermodynamics of acetate salt solutions10. A number of ab initio calculations of ion coordination with amino acids in gas-phase were conducted previously13–15; however solvent effects are significant and should be taken into account10,16,17.
To better understand the molecular details of ion pairing on protein-protein interactions, the spatial distribution of ion positions is of interest. Here we present a molecular dynamic study of the spatial distribution of sodium and potassium coordinated with zwitterionic glycine in a concentrated water ionic solution.
Molecular dynamic (MD) simulations were conducted in GROMACS package (version 4.6.7)18. Simulation systems contained one zwitterionic glycine molecule (as at pH 7 it is the most probable glycine form in solution), 33 cations, 33 chloride anions and about 800 water molecules in a cubic periodic box with 3 nm sides, corresponding to a 2 M salt solution. Equilibration of 10 ns preceded 500 ns production MD for each system with constant number of particles (N), constant pressure (P) and temperature (T) conditions – in NPT ensemble. Temperature at 300 K and pressure at 1 bar were maintained with Nose-Hoover thermostat19,20 and Parrinello-Rahman barostat21. Electrostatics PME method was used22 with grid spacing of 0.12 nm and 1.0 nm cutoff, the same as for van der Waals interactions. For zwitterionic glycine, parameters were from OPLS-AA force field23, all bonds were constrained with LINCS algorithm24 (for more details on parameters see run input files available in Dataset 1). Parameters for cations were obtained from 25, for chloride from 26 and a TIP3P water model was used27. Radial distribution functions were calculated with bin width of 0.004 nm using g_rdf utility of GROMACS package. Spatial distribution were calculated with g_spatial GROMACS utility after least square fit of heavy atoms of glycine molecule from each frame to the position of starting MD structure.
Two systems were investigated, each consisted of one glycine dissolved in explicit water with sodium and chloride ions, or potassium and chloride ions. Radial distribution functions (RDF) of Na+ or K+ with respect to oxygen atoms or carbon atoms of glycine carboxylate group were calculated and are plotted in Figure 1 The O-Me+ RDF for both studied ions shows several coordination shells with a pronounced first maximum that is considerably higher for sodium, which lies in agreement with previous studies indicating a superior Na+ affinity6,7,10,12,17. Analysis of C-Me+ RDF, however, is not so common in the literature. C-Me+ RDFs are plotted in Figure 1B, and show two sharp peaks for sodium ions at 0.28 and 0.34 nm, as well as two weaker separate, but distinct peaks for potassium at 0.32 and 0.36 nm. This figure indicates that there are two favorable coordination states of cations with carboxylate groups which both contribute to the single first peak of O-Me+ RDFs.
Figure 2 shows the iso-density surfaces of sodium or potassium ions calculated around glycine and explicitly reveals these coordination states. One sees the medial (m) coordination state equidistant from the oxygen atoms of the carboxylate group, and the lateral (l) state consisting of the two regions being closer to one of the two oxygen atoms. The asymmetry that is seen in the shape of the (l) state regions, including a bridge connecting the (m) and (l) in the case of K+, occurs due to positively charged NH3 group and overall conformational flexibility of glycine. Density levels on the spatial distribution for sodium considerably exceed that for potassium, and during the simulations we occasionally observed only for sodium glycine coordinated with two ions in (m) and (l) states simultaneously (see Figure 3). Distances depicted in Figure 3 clearly illustrate that the (m) state is closer to the C of carboxylate group and corresponds to the first peak on C-Me+ RDF (0.28 nm for Na+). The (l) state corresponds to the next peak (0.34 nm after minimum at 0.3 nm for Na+), while both states belong to the same peak of O-Me+ RDF (0.23 nm for Na+). In sodium simulation, glycine exists with Na+ in (m) coordinated state for 21% of the observation time and with Na+ solely in (l) coordinated state for 30%. For potassium simulation, we obtained 8% and 18% of time for (m) and (l) coordinated states, respectively.
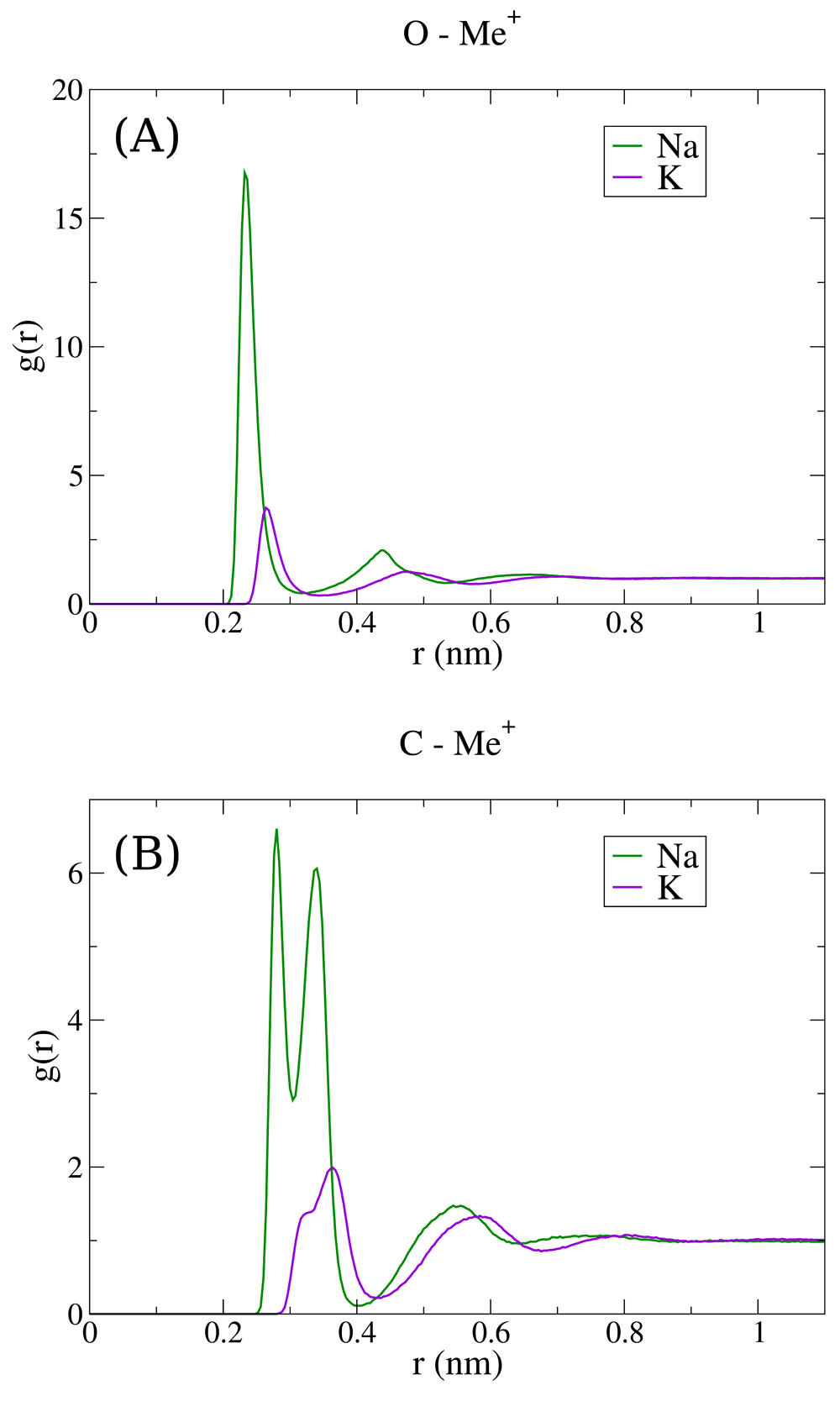
(A) O-Me+ RDF, (B) C-Me+ RDF. Green, sodium; violet, potassium.
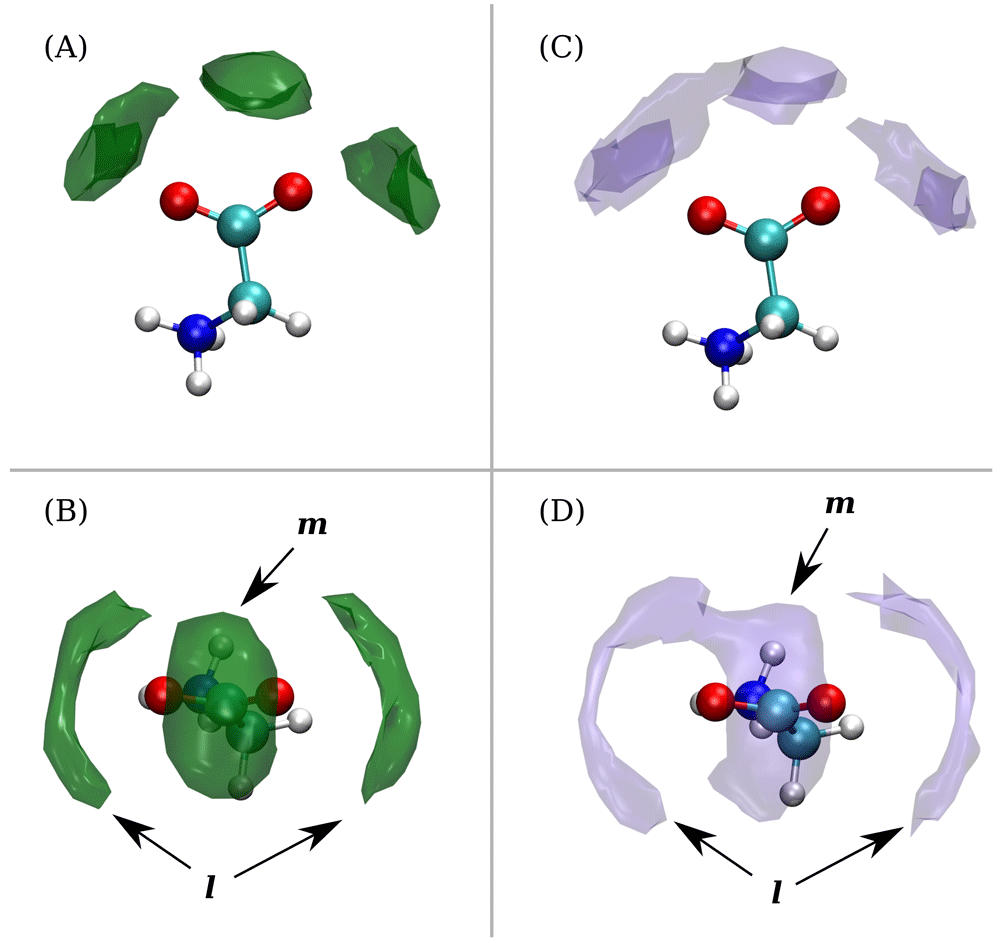
Iso-density surfaces around glycine are shown for Na+ (A and B) and K+ (C and D). Note that the density value (cutoff) for Na+ is much higher than for K+.
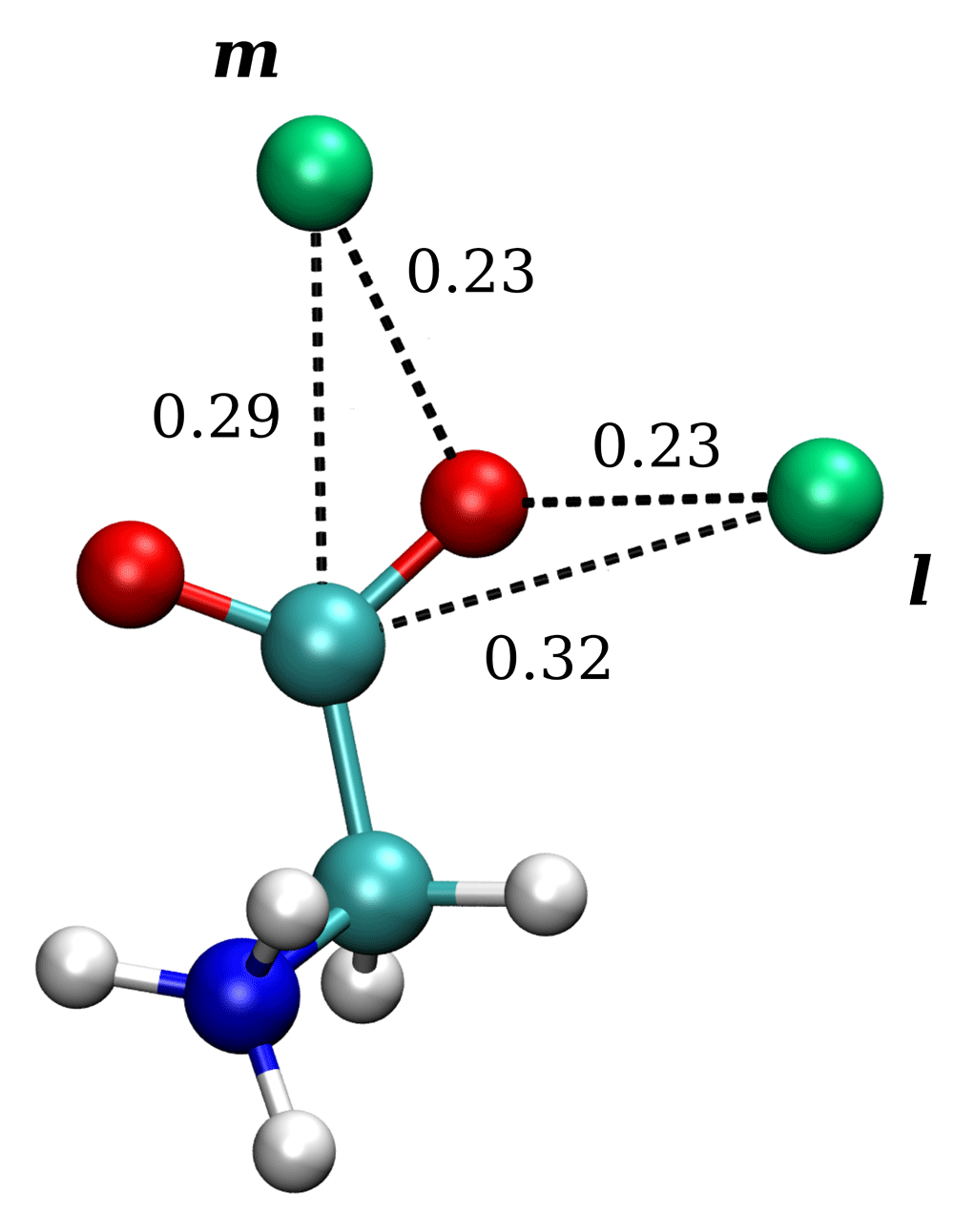
Distances are given in nanometers. Green, sodium atoms; red, oxygen; gray, carbon; water molecules not shown.
We demonstrated that contact ion pair of carboxylate group with Na+ or K+ splits into distinct, well occupied, (m) and (l) coordination states. The effect may be of interest in studies devoted to ab initio calculations and in the interpretation of X-Ray absorption data, as they account for (m) coordination state only6–8. Coordination with ions is thought to be crucial in the first stage of abiogenic peptide polymerization process28 and therefore, the observed differences in sodium and potassium behavior are important for research into primary abiogenic peptide synthesis conditions.
Dataset 1: Run input parameters for production MD in GROMACS version 4.6.7. doi, 10.5256/f1000research.10644.d14976429
Dataset 2: Input typologies and equilibrated structures for production MD (in zipped file). doi, 10.5256/f1000research.10644.d14976530
Dataset 3: MD trajectories (.xtc) and input files (.tpr) for NaCl and KCl systems (in zipped file). Positions of water molecules are not included. doi, 10.5256/f1000research.10644.d14976631
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)