Keywords
nitric oxide, neuronal nitric oxide, cardiac protection, heart disease
nitric oxide, neuronal nitric oxide, cardiac protection, heart disease
Nitric oxide (NO) is an essential molecule that plays fundamental roles in maintaining cardiovascular functions in animals and humans1–5. The NO that exerts biological functions in the myocardium can be acquired through exogenous sources or is produced from the endogenous endothelial and neuronal NO synthases (eNOS and nNOS, respectively, which are constitutively expressed in the myocytes) and from inducible NOS by inflammatory cytokines following infection4–6 (Figure 1). In the last few decades, our understanding of the detailed mechanisms, the effects of NO on myocardial functions, and the roles for NOSs in diseased hearts has improved. Comprehensive approaches have been undertaken to achieve this outcome, including manipulation of the upstream and downstream effectors of NOSs (pharmacologically or genetically modified NOS regulation and viral infections of specific NOS genes), supplementation of NO mimetics (for example, exogenous NO donors or NO substrates), and systematic detection of plasma and tissue NO4,5,7,8. Nevertheless, the practical implications of NO and its precursors or regulators in translational and therapeutic strategies in cardiovascular diseases are hampered because of the complex nature of NO and the array of downstream signalling cascades and effectors in the myocardium. In the initial part of this review, I will provide a systematic overview of the sources and post-transcriptional modification of NO in the myocardium; the latter part of the review will focus on recent advances of nNOS-targeting proteins and responses in the heart under stress. Ultimately, I will delineate the prospect for using the therapeutic platform of nNOS and NO to target human heart diseases.
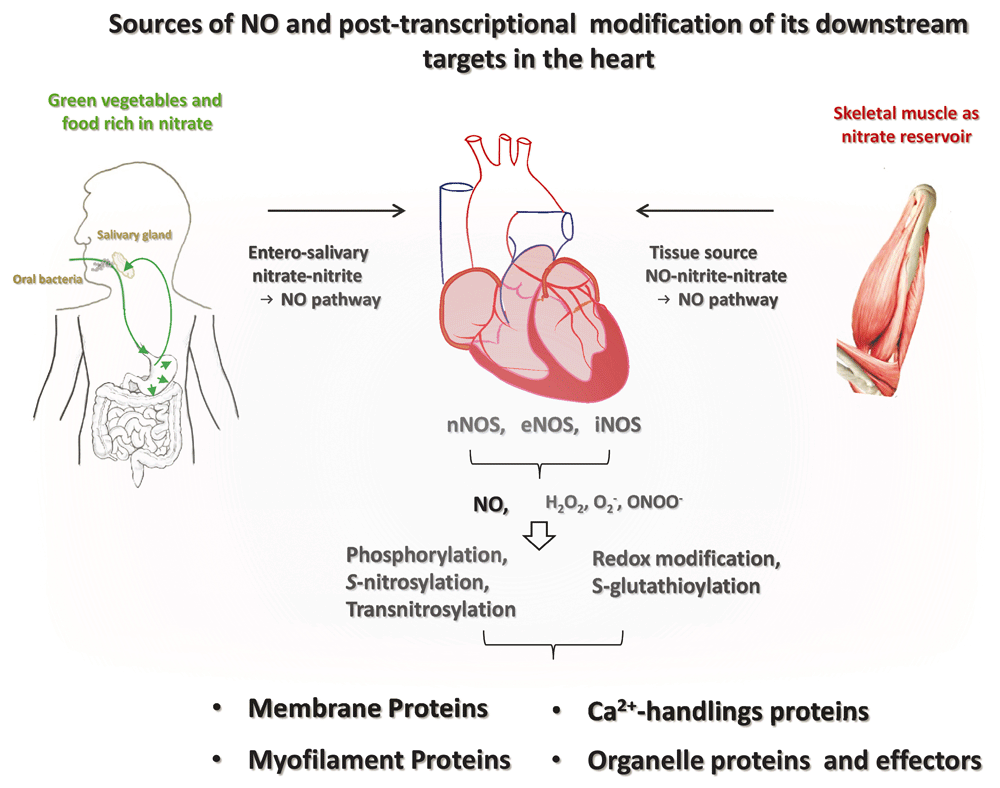
Both exogenous sources (nitrate-rich vegetables and food through the entero-salivary nitrate–nitrite–NO pathway and skeletal muscle nitrate → NO pathway) and endogenous sources (neuronal nitric oxide synthase [nNOS], endothelial NOS [eNOS], or inducible NOS [iNOS]) determine the bioavailable NO in the myocardium. NO regulates downstream targets through soluble guanylate cyclase (sGC)/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG)-dependent phosphorylation, S-nitrosylation, and transnitrosylation. Alternatively, NOS-derived radicals and NO-related derivatives (H2O2, O2–, and peroxynitrite [ONOO–], etc.) affect downstream effectors through the oxidation and S-glutathionylation. As such, NO regulates membrane proteins, Ca2+-handling proteins, membrane proteins, and organelle effectors in the cardiac myocytes.
It is generally acknowledged that NO is derived from the classic L-arginine–NOS–NO pathway. In fact, NO that exerts functions in the myocardium may also be acquired from the alternative source of NO, the nitrate–nitrite–NO pathway (Figure 1). Nitrate (NO3−) in various types of green leafy vegetables and food9–11 is taken up into the plasma to become a reliable reservoir and the stable precursor of NO (the half-life of nitrate in the plasma is 5–6 hours). Nitrate from this source is actively taken up by the salivary gland, is secreted in concentrated form in the saliva (about 10-fold that in the plasma), and is subsequently reduced to more active nitrite (NO2−) in the oral cavity by nitrate reductases of commensal bacteria (entero-salivary NO pathway). Nitrite is reduced to NO in the acidic environment of the stomach and is absorbed into the blood in the upper gastrointestinal system (the half-life of nitrate in the plasma is about 30 minutes), while the rest mixes with the nitrite formed from nitrate derived from endogenous NOS-produced NO. Various enzymes and proteins are known to be involved in the NO metabolite cycle and nitrite’s reduction to NO, including xanthine oxidase12,13, deoxyhaemoglobin and deoxymyoglobin14–16, neuroglobin17, respiratory chain enzymes18, cytochrome P45019, aldehyde oxidase20, carbonic anhydrase21, and NO synthase22. Overall, about 25% of nitrate undergoes re-uptake by the salivary gland and produces functional NO in the circulation; the rest of the nitrate is eventually excreted in the urine. The amount of NO from exogenous sources can be as high as the amount that is produced from NOSs in the tissues (with enough daily consumption of green leafy vegetables or food, nitrite intake varies from 0 to 20 mg/day23), indicating the importance of this pathway in supplementing local NO in the tissue. Notably, unlike NO from the L-arginine–NOS–NO pathway, food-derived functional NO is oxygen independent10,11. Accordingly, NO from this source becomes more important in ischaemic or hypoxic conditions, such as myocardial infarction, hypertrophy, and heart failure.
NO or nitrite can undergo an oxidative process via oxyhaemoglobin or oxymyoglobin to produce stable nitrate, which can be reduced back to nitrite and NO by molybdopterin-containing mammalian nitrate reductases, such as xanthine oxidoreductase or aldehyde oxidase10,11. Therefore, there is a constant recycling of NO precursors, metabolites, and NO that maintains exogenous NO in the human body. The respective contributions of the endogenous versus exogenous NO to intracellular signalling and function in healthy and diseased hearts in vivo remain to be revealed.
A number of organs or tissues (for example, neurons, liver, heart, skeletal muscle, kidney, arteries such as the aorta, and endothelium) are the active sites for NO production from constitutive NOSs. Recently, it has been shown that skeletal muscle is a dynamic nitrate reservoir that increments plasma nitrate and nitrite because of the abundance of the tissue in the mammalian body24. nNOS in the skeletal muscle contributes to the supply because it is the only isoform in the skeletal muscle25. However, the proportions of NO from the specific sources that contribute to the bioavailable NO in the myocardium remain undetermined.
Classically, eNOS is the primary isoform of NOS that plays important roles in NO regulation of physiological functions in the majority of tissues, including the heart4,5,7,8. In the cardiac myocyte, eNOS is located in spatial microdomains of the plasma membrane (caveolae and lipid rafts), Golgi apparatus, nucleus, and mitochondria8,26. eNOS displays the highest activity at the plasma membrane, followed by outer membranes of the cis-Golgi and low activity in the cytosol, nucleus, and mitochondria26,27; therefore, localisation is the main determinant of eNOS activity for specific biological functions. Conversely, mis-localisation of eNOS has been shown to reduce its capacity to generate NO in intact cells26–28. Post-translational cysteine palmitoylation (Cys15 and Cys26) or N-myristoylation at Gly2 of eNOS, catalysed through Asp-His-His-Cys motif-containing palmitoyl acyltransferases, is critical in locating eNOS to the membrane for optimising its activity28,29.
A number of alternatively spliced eNOS variants have been identified: specifically, eNOS lacking exons 20 and 2130 or three splice variants of eNOS containing novel 3' splice sites within intron 1331. These alternative splicing variants produce truncated isoforms of eNOS with maintained30 or diminished31 NO-producing activity. Reduced eNOS activity has been shown in the heterodimer of eNOS (the splice variants with the full-length eNOS)31, although the existence of the splice variants of eNOS and their functional impact in the myocardium are unclear. In diseased hearts, the protein expression and the activity of eNOS are known to be downregulated32–34 or uncoupled35. These results suggest that maintaining eNOS protein and activity in its “coupled form” is beneficial by preventing the early stage of disease progression in the heart.
Recent consensus is that nNOS is the isoform that plays the principal role in cardiac physiology and pathology because nNOS is expressed in all parts of the heart, including the autonomic nervous system innervating the heart, aortic and pulmonary arteries, coronary artery, and the atrial and ventricular myocardium7,8. As such, nNOS is well placed to fill essential roles in modifying sympathetic and parasympathetic tones, controlling heart rate, delivering essential nutrients through coronary arteries, and regulating myocardial contractility. In the myocardium, nNOS is predominantly localised in the sarcoplasmic reticulum (SR)6 and is involved in the Ca2+ handling processes of cardiac excitation-contraction coupling7,8. In addition, nNOS interacts with α-syntrophin through the scaffolding protein postsynaptic density-95 (PSD95) via the PSD-95/Discs large/ZO-1 homology domain (PDZ domain) and forms a multi-protein complex with the plasma membrane Ca2+ pump (PMCA) and voltage-gated Na+ channel (Nav1.5)36. In addition, nNOS binds to its PDZ-binding motif to direct nNOS to the subcellular compartments, as is the case of nNOS in the nucleus37, which regulates the transcription and activation of the elements required for oxidative phosphorylation and mitochondrial biogenesis37. Recently, we have shown that nNOS is upregulated in the myocardium from the early stage of disease progression (e.g. hypertension34) and facilitates lusitropy through myofilament Ca2+ desensitisation34.
Until recently, most of the responses of nNOS were attributed to nNOSα or nNOSμ7,8. However, the existence of various splice variants of nNOS (nNOSβ, nNOSγ, and nNOS2) suggests that splice variants of nNOS may be involved in producing NO and regulating contractile function in the heart. Very recently, we have presented novel evidence to show that nNOSβ, which does not possess the PDZ domain, is expressed in the myofilament fraction of cardiac myocytes from the hearts of healthy and hypertensive rats38. These results indicate that nNOSβ may play important roles in cardiac myofilament. On the other hand, it has been documented in skeletal muscle that nNOSβ is functionally expressed in the Golgi apparatus and mediates myofilament regulation during exercise25. A comprehensive understanding of nNOS and its splice variants in the organelles and their roles in cardiac function and protection in the healthy and diseased hearts remains to be explored.
Notably, the co-existence of eNOS and, nNOS and their splice variants in the myocardium and their translocation, transcription, and post-translational modifications underlie the complex scenario of NO in the heart7,8,39, more so under pathological stress. This is represented by a contrasting tendency in protein expression and activities of eNOS and nNOS in the failing myocardium or in the hypertensive heart; that is, eNOS protein expression is reduced significantly, whereas nNOS protein expression and activity are increased32–34,40. Furthermore, nNOS in the SR translocates to the caveolae to protect the myocardium from Ca2+ overload and oxidative stress7,33,41. Intriguingly, both eNOS and nNOS affect intracellular Ca2+ handling in the myocytes, and eNOS mediates spontaneous Ca2+ sparks and enhanced Ca2+ transients in cardiac myocytes in response to increased preload (mechanical stretch)42. Conversely, nNOS (but not eNOS) mediates the afterload-induced spontaneous Ca2+ sparks43. Spatial redistribution of NOSs is associated with both the changes of their activity and the shifting of the primary targets that underlie the mechanisms of myocardial function under stress. In essence, the translocation of nNOS may be beneficial in maintaining its activity to exert cardiac protection.
It is generally accepted that S-nitrosylation (or S-nitrosation) and soluble guanylate cyclase (sGC)/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG)-dependent phosphorylation are the predominant mechanisms that mediate the effects of NO in biological systems (Figure 1). The former mechanism involves post-translational modification of a thiol group in proteins by NO (transferring NO to cysteine residues, -SNO), and the latter implicates PKG-dependent phosphorylation of serine residues of the target proteins. S-nitrosylation is explicitly initiated by NO, but dinitrogen trioxide (N2O3), the nitrosonium ion (NO+), peroxynitrite (ONOO−), and SNO proteins are also able to deliver NO to the cysteine residues of the target proteins44,45. Protein-protein transfer of NO (trans-S-nitrosylation) is now known to represent one of the most important mechanisms of NO46 (Figure 1). In this process, the SNO “donor” proteins are referred to as nitrosylases. Trans-S-nitrosylation possesses advantages for effective interactions between proteins47. Furthermore, transnitrosylation is important when NO bioavailability is limited in an oxidative and/or nitrosative stress environment, such as during ischaemic reperfusion. S-nitrosylation can be terminated by the action of denitrosylases (for example, S-nitrosoglutathione reductase and thioredoxin), with NADH and NADPH serving as electron donors to regenerate glutathione and thioredoxin48,49.
Various types of proteins are targeted by NO, which in turn triggers an array of signalling cascades depending on the properties of the target proteins, e.g. inhibition of protein phosphatase 2A/protein phosphatase 1 by NO leads to protein kinase A (PKA) and Ca2+-calmodulin-dependent kinase II-dependent phosphorylation of downstream effector proteins such as phospholamban (PLN)50, whereas sGC activation by NO in the myocardium of hypertensive rats causes cGMP/PKG-dependent phosphorylation of cTnI and cMyBPC34. Conversely, phosphodiesterase 5 (PDE5) activation by NO/sGC/cGMP/PKG limits cytosolic cGMP, a negative feedback mechanism of NO regulation of cGMP in cardiac myocytes51. In addition, by targeting cardiac oxidases, such as xanthine oxidoreductase52, NADPH oxidase53,54, and mitochondrial reactive oxygen species (ROS) production55, nNOS-derived NO controls intracellular oxidative status and ROS-dependent downstream effects in the myocardium. Cysteine residues are the targets of ROS to cause S-glutathionylation in the proteins56,57; therefore, S-nitrosylation by NO may “block” critical cysteine residues from irreversible oxidation under the conditions, such as increased oxidative stress. Consequently, post-transcriptional modifications downstream of NO change the effector proteins, altering their localisation, binding partners, activity, and, ultimately, function.
nNOS has also been demonstrated to produce H2O2 in the endothelium of large arteries, such as the aorta, and H2O2 mediates endothelium-dependent vascular relaxation58,59. Conversely, impairment of endothelial nNOS-derived H2O2 has been shown to worsen endothelial dysfunction in both atherosclerosis60,61 and hypertension62, indicating a protective role of nNOS-derived H2O2 in the vasculature. Similarly, both eNOS-derived NO and nNOS-derived H2O2 contribute to acetylcholine stimulation of vasodilatation59 by regulating similar downstream protein kinases and phosphatases63–65. In contrast, uncoupling of eNOS and nNOS (secondary to the deficiency of L-arginine, tetrahydrobiopterin [BH4] oxidation, or S-glutathionylation52,66–68) results in the production of superoxide (O2−) instead of NO; under such conditions, eNOS and nNOS become the sources of oxidative stress for pathological progression in the myocardium.
Taken together, the mechanisms mediating the effect of NO are complex. S-nitrosylation, transnitrosylation, and sGC/cGMP/PKG-dependent phosphorylation provide major post-transcriptional modifications of NO. By producing H2O2, O2−, and the NO derivatives, NOSs also function as the upstream regulators of redox-dependent signalling.
nNOS exerts its cardiac protection through the regulation of ion channels, modulating abnormal Ca2+ homeostasis, mitochondrial function, and signalling pathways during pathological progression7,8 (Figure 1). To fulfil the effects on cardiac electrophysiology and intracellular Ca2+ homeostasis, nNOS regulates key ion channels and Ca2+-handling proteins that participate in the process of electrical activity and excitation-contraction coupling of cardiac myocytes. In particular, nNOS has consistently been shown to reduce Ca2+ influx through the L-type Ca2+ channel (LTCC)69, and its effect is potentiated in cardiac myocytes of female mice following post-ischaemia/reperfusion and significantly reduces ischaemia/reperfusion injury41. In support of this, nNOS increases the vulnerability of the LTCC for Ca2+-dependent inactivation in hypertensive cardiac myocytes70 where intracellular Ca2+ transient is increased secondary to nNOS-dependent myofilament Ca2+ desensitisation34. The effect of nNOS on the LTCC can be mediated by both S-nitrosylation and cGMP/PKG-dependent phosphorylation41,71,72. Modulation of the LTCC by nNOS may prevent excessive intracellular Ca2+ loading in cardiac myocytes under pathological threat. S-nitrosylation of the ryanodine receptor (RyR) by nNOS has been implicated in reducing diastolic Ca2+ leak73, increasing RyR open probability, and increasing contraction in cardiac myocytes74. Therefore, nNOS protects against arrhythmogenesis by modulating Ca2+ transients75,76. On the other hand, greater nNOS activity at the plasma membrane (subsequent to dissociation from the PMCA-containing complex) induces greater Na+ influx through voltage-gated sodium channels (Nav1.5) via S-nitrosylation and enhances the susceptibility of the myocardium for long QT and arrhythmias38. Potassium channels are also potential targets of nNOS through S-nitrosylation and/or cGMP/PKG-dependent phosphorylation77–79, which may play important roles in the regulation of cardiac electrophysiology and mechanical function in both healthy and diseased hearts.
nNOS-derived NO, or the formation of ONOO−, can induce S-nitrosylation of the SR calcium ATPase (SERCA) both under basal conditions and with stimulation76,80 (for example, myocardial infarction). Inhibition of nNOS reduces S-nitrosylation of SERCA at basal level, and this is associated with reduced Ca2+ uptake in the SR and decreased relaxation80. However, the functional significance of this regulation under disease conditions remains to be determined. Alternatively, SERCA activity can be increased by nNOS via PKA-dependent phosphorylation of PLN secondary to nNOS-dependent inhibition of protein phosphatase 2A activity in left ventricular (LV) myocytes from normal mice50. A recent report has shown that beta-adrenergic stimulation induces S-nitrosylation of PLN and increases its pentamerisation and the activation of SERCA. Whether the source of NO for the S-nitrosylation is from nNOS is not revealed in the study. However, nNOS-dependent PLN pentamerisation following beta-adrenergic stimulation are shown not to affect basal and beta-adrenergic phosphorylation of PLN. This is important because the results confirm that nNOS, either through S-nitrosylation under β-adrenergic stimulation or through phosphorylation secondary to the inhibition of protein phosphatases, promotes SERCA activity and exerts positive lusitropy in the myocardium. In addition, these results emphasise that S-nitrosylation and phosphorylation work in concert to mediate the effects of nNOS on cardiac function.
On the other hand, phosphorylation of PLN (Ser16) is increased by nNOS through a cGMP/PKG-dependent mechanism independent of scavengers of ONOO−, O2−, or PKA in cardiac myocytes stimulated by angiotensin II (Ang II) where nNOS is upregulated53. Furthermore, phosphorylation of PLN (Ser16) is increased in Ang II-induced hypertensive rat ventricular myocytes, but this response is independent of nNOS or cGMP/PKG signalling and exerts little effect on nNOS facilitation of myocyte relaxation34. These results suggest that the modes of post-transcriptional modification that underlie the specific effects of nNOS are highly dynamic, and this may optimise its regulation of the downstream target proteins under various stimuli, including pressure overload.
A recent study from our own group has shown that nNOS-derived NO increases cGMP/PKG-dependent phosphorylation of cardiac troponin I (cTnI-Ser23/24) and cardiac myosin binding protein C (cMyBPC-Ser273) and promotes myocyte relaxation in the hypertensive heart through cGMP/PKG-dependent myofilament Ca2+ desensitisation34, indicating the involvement of myofilament proteins in nNOS-dependent responses in hypertensive myocardium. In fact, isobaric tag for relative and absolute quantitation (iTRAQ)-based quantitative proteomic analysis shows that nNOS affects the phosphorylation of almost 20 myofilament proteins in LV myocytes from the healthy heart and a similar number of distinct proteins in the hypertensive heart38. These results indicate that myofilament proteins are the potential targets of nNOS that mediate faster relaxation in cardiac myocytes to reduce the mechanical load of the myocardium in hypertension. This is consistent with previous findings that exogenous NO donors facilitate myocardial relaxation via sGC and cGMP/PKG-dependent phosphorylation of cTnI and myofilament Ca2+ desensitisation81. A recent report has demonstrated that NO mimetics (S-nitrosocysteine) reduce myofilament Ca2+ sensitivity and myocardial contractility by inducing the S-nitrosylation of a number of myofilament proteins including actin, myosin, cMyBPC, and troponin C (cTnC)82. More directly, both TnC-Cys35 and TnC-Cys84 are S-nitrosylated by beta-adrenergic stimulation and TnC-Cys84 is shown to be responsible for reduced myocardial Ca2+ sensitivity in normal hearts. In contrast, S-glutathionylation of myofilament proteins in the hypertrophic myocardium increases myofilament Ca2+ sensitivity and impairs relaxation83,84. These results strongly indicate that phosphorylation and S-nitrosylation (as well as oxidation) of myofilament proteins are the fundamental mechanisms that mediate the effects of nNOS in normal and diseased hearts.
nNOS is regarded as the potential isoform that is expressed in the mitochondria to actively regulate cardiac metabolism85. NO inhibits cytochrome c oxidase (complex IV) activity by competing with O2 and inhibits electron transfer of complex III (between cytochrome b and c) or NADH-dehydrogenase function at the level of complex I and increases mitochondrial production of O2−. Consequently, NO inhibits the mitochondrial respiration chain and reduces mitochondrial oxygen consumption86–91. As such, NO has generally been acknowledged as the negative regulator of mitochondrial activity and energy metabolism. This is seemingly counterintuitive to the cardiac protection of nNOS in the diseased heart or in the heart under stress because of the consensus that nNOS exerts protective roles. Nevertheless, conditional overexpression of nNOS in the myocardium has been associated with increased nNOS in the mitochondria and attenuation of mitochondrial ROS production and a reduction in oxidative stress following myocardial infarction55. Although it remains to be confirmed, the modulation of oxidative stress by endogenous nNOS in diseased hearts can be a potential protective mechanism.
Emerging evidence shows that nNOS-derived NO plays essential roles in mitochondrial biogenesis92,93 to maintain or increase mitochondrial integrity and activity. For example, nNOS has been shown to be redistributed to the nucleus via α-syntrophin through its PDZ domain in a variety of cells, including myocytes37,94. Increased S-nitrosylation of nuclear proteins, including cAMP response element-binding protein (CREB), in turn, interacts with the promoter of the gene encoding peroxisome proliferator-activated receptor γ co-activator (PGC)-1α promoter, an essential component for mitochondrial biogenesis and nuclear respiratory factor 1 and mitochondrial transcription factor A37. S-nitrosylation of nuclear proteins has also been ascribed to the trans-S-nitrosylation activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH)95.
Additionally, NO has been implicated in cardiac energetics by affecting carbohydrate metabolism both within and outside of mitochondria. For example, NO stimulates glucose transport by activating upstream signalling pathways that result in increased amounts of the glucose transporter GLUT4 at the cell surface96. Accordingly, inhibition of NOSs reduces the uptake of glucose and ATP production in skeletal muscle, both under basal conditions and during physical activity96–99. Moreover, NO has been implicated in the inhibition of the glycolytic enzyme GAPDH by means of S-nitrosylation100,101.
It should be noted that both nNOS and eNOS are required for mitochondrial activity, including biogenesis in many types of cells, such as myocytes92,102,103. A typical example is that bradykinin inhibits mitochondrial oxygen consumption via eNOS in myocardial tissue104 and that nNOS-derived NO remains unaffected. However, in the absence of nNOS, reduced NO bioavailability secondary to increased xanthine oxidase-derived O2− limits the effect of eNOS in controlling mitochondrial oxygen consumption105, indicating the interplay between eNOS and nNOS in the regulation of mitochondrial activity and cardiac metabolism. Detailed mechanisms of the interaction between eNOS and nNOS in mitochondria and its functional relevance in healthy and diseased hearts, however, remain to be determined.
Because of the importance of NO and its significance in the cardiovascular system, approaches that manipulate the bioavailability of NO in the myocardium are essential therapeutic strategies for the better treatment of cardiovascular diseases (CVDs). In fact, nitroglycerin, an organic nitrate that releases NO, has been used clinically in the treatment of CVD for more than 150 years (despite the fact that the effective molecule for the response, NO, was identified in the late 1970s1). Enhanced acknowledgement of the mechanistic insights into NO signalling, exogenous versus endogenous NO sources, the maintenance and the degradation of NO, and the properties of NOSs as well as modern technology enables novel approaches to increase NO bioavailability in target tissues for the desired responses. In principle, enhancement of NO and its signalling can be achieved through three routes: increase exogenous and endogenous sources to promote NO production, reduce NO metabolism/degradation, and stimulate downstream signalling of NO.
A number of strategies are used to promote NO formation. For example, inhaled NO is registered to be applied to newborn babies with persistent pulmonary artery hypertension106,107 to support ventilation-perfusion match and to prevent systemic ischaemia. Nitrite can be similarly applied; in fact, the effectiveness of oral, inhaled, and intravenous nitrite on a number of CVDs (such as pulmonary artery hypertension [PAH], peripheral vascular diseases, myocardial infarction, and cerebral vasospasm after subarachnoid haemorrhage) are under study with promising prospects on some occasions108–110. Delivering organic and inorganic nitrate and nitrite to amplify systematic or local NO through nitrate–nitrite–NO and the nitrate–nitrite–NO–fatty acid pathways are probably the most active area under investigation experimentally and in the clinic10. So far, a number of putative precursors of NO (nitroxyl [HNO], S-nitrosothiols, sodium nitrite, sodium nitrate, nitrated fatty acids, and nitrate from beetroot juice and green leaves, such as spinach, etc.) have been identified and are under development. Dietary consumption of NO precursors is a cheap, safe, and effective way of nitrate delivery; programming of a suitable diet regime for vulnerable populations will be important to reduce the cardiovascular risks as well as the economic burden on national healthcare systems. The relationship between the daily consumption of nitrate and cardiovascular events is noticeable. For example, high fruit and green vegetable intake in a Japanese population historically known to have low rates of CVD (daily consumption of nitrate >1,100 mg/60 kg) is associated with greater circulating nitrate and nitrite111 compared to those in the US and Europe, where average daily nitrate consumption ranges from 40–100 mg and 30–180 mg, respectively, and the rates of CVD are high112,113. Moreover, the consumption of “healthy” fats, as in the Mediterranean diet, in the form of unsaturated fatty acids such as oleic and linoleic acid (to form nitrated fatty acids), is beneficial in preventing the development of CVD and reduces the risk factors114. Notably, nitrite reduction to NO preferentially occurs in the presence of hypoxia and acidosis, during physical exercise, at the time when cardiac muscle needs NO the most.
Alternatively, supplementation of NO substrates, e.g. arginine, L-citrulline, and BH4 (a co-factor of NOS), and inhibition of arginase and asymmetric dimethylarginine (an endogenous NOS inhibitor) are the necessary strategies to increase NO through promoting NOS activity11. Statins and nebivolol or carvedilol (new-generation beta1-adrenergic receptor blockers) exert anti-adrenergic responses via the stimulation of the beta3-adrenergic receptor and increasing NOS production of NO115–118. Targeting NOS is advantageous in mediating the specific downstream signalling of NOSs in the compartments.
Attempts to prevent NO reduction and inhibition of NOS uncoupling are also important in maintaining or increasing cytosolic NO. Decreasing the formation of ROS using blockers of angiotensin-converting enzyme (ACE), angiotensin I type 1 receptor (AT1R), or NADPH oxidases (NOXs) or reducing ROS by using antioxidants and scavengers are the putative mechanisms to reduce NO “sink” and therefore maintain or increase NO level11. However, the complex NOX isoforms and the redox–nitrosol network weaken the effectiveness of the developed drugs in clinical use. Indeed, our recent results indicate that NOX and ROS are upstream regulators of cardiac nNOS protein and activity downstream of Ang II and AT1R119. Furthermore, Ang II type 2 receptor (AT2R) mediates Ang II enhancement of nNOS protein expression via ROS activation of eNOS activity119. As such, it is wrong to simply assume that NO level can be increased by using ACE and AT1R inhibitors. The development of selective NOX inhibitors and specific ROS-manipulating drugs that do not affect NOS protein should be pursued, and the effect of NOX and ROS on nNOS protein expression in the myocardium should be taken into consideration.
Stimulation of the downstream signalling pathway of NO is an improved strategy to target the effector proteins directly, bypassing the complex scenario of NOSs and the redox–nitrosol network. Synthetic benzyl indazole compound YC-1, the oral sGC stimulators riociguat and vericiguat, and atrial, brain, and C-type natriuretic peptides are in use to increase cellular cGMP11. Inhibition of a negative regulator of cGMP, PDE5 (e.g. using sildenafil), is another therapeutic approach to stimulate cGMP/PKG signaling120–122. Stimulation of the PKG-dependent pathway has been shown to exert potent protective effects in a wide array of cardiovascular disease models, including hypertension, PAH, heart failure, haemolytic anaemia, and infarct-reperfusion injury120–124. However, the application of the drugs in a large cohort of patients with CVD show minor responses to the treatment, suggesting further investigation on cGMP enhancers is still needed.
Primary S-nitrosothiols (RSNOs) are the endogenous NO carriers and donors and have emerged as platforms for the localised delivery of NO, which mediates S-nitrosation-dependent mechanisms125. S-nitrosoglutathione (GSNO) is one of the main RSNOs and is a central intermediate in the formation and degradation of cellular S-nitrosothiols with potential therapeutic applications125. So far, GSNO has not been implied in pharmaceutical composition owing to the fast decomposition in aqueous solutions. To sustain the bio-available NO donors and optimise their kinetics and to control the delivery of NO to targeted tissues or proteins, various biomaterial-based carriers (microparticles and nanoparticles) are under development126.
Taken together, a number of validated ways have been developed to increase systemic and local NO levels and are promising in mediating the beneficial effects in CVD. However, in order to translate the research innovations into the application to a large population, more research is necessary, with special attention to the specificity and effectiveness, i.e. nitrate/nitrite regime in the diet and strategies of increasing nNOS (as well as eNOS) and improving NO-effector interactions in CVD settings. NO-dependent therapy holds promise as a therapeutic platform for CVD in humans.
Our understandings of the NO and NOSs that regulate myocardial contraction, relaxation, and pathological signalling are advanced, but the dynamic paradigm in the myocardium under stress is not clearly presented. NO from both exogenous and endogenous sources supply the bioavailable NO in the myocardium, and the level and effectiveness of NO is determined by multiple regulation mechanisms including daily consumption of NO precursors, nitrate from skeletal muscles, NO production through the entero-salivary NO pathway (oral and gut microbiome function as essential regulators) and from NOSs as well as redox environment and the nature and the abundance of target proteins. In general, NO regulates downstream effector proteins through three mechanisms (sGC/cGMP/PKG-dependent phosphorylation, S-nitrosylation, and transnitrosylation) and the numbers and types of effectors regulated by NO are diverse. As such, modification of these effectors by NO subsequently triggers an array of signalling cascades that lead to different physiological and pathological consequences. By and large, NO and its downstream signalling pathway exert potent cardiovascular protection; however, translational research of NO and NOS that are applicable for CVD and therapeutic efficiency using an NO-dependent regime are still far from satisfactory11. Further research needs to be carried out aiming to increase the specificity of NO-effector interaction at the site of relevant proteins. Better design of a dietary program and the promotion and targeting of nNOS and eNOS in specific locations with the effector target(s) of importance will maximise the effectiveness of NO application in cardiovascular physiology and pathology.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)